



1. Введение
Сравнительный анализ белков является важнейшим шагом при изучении структурно-функциональных взаимосвязей. В процессе эволюции изменение аминокислотной последовательности происходит с большей скоростью, чем изменение структуры [2]. Это приводит к тому, что ферменты, произошедшие от одной предковой формы в результате дивергентной эволюции и претерпевшие значительные функциональные изменения в условиях естественного отбора, не обладают сходством по последовательностям, достаточным для сравнительного анализа [2; 7]. В противоположность выравниванию аминокислотных последовательностей белков выравнивание структур в первую очередь основано на геометрическом сравнении положения аминокислот в пространстве, а не на сопоставлении их биохимических особенностей. В связи с этим при изучении структурно-функциональных взаимосвязей между дальними эволюционными родственниками выравнивание третичных структур представляется значительно более информативным и точным, чем выравнивание аминокислотных последовательностей. В настоящий момент в банке данных трехмерных моделей белков PDB насчитывается более 85 000 записей, число которых ежегодно растет [1]. Доступность этой информации открывает новые перспективы для биоинформатического структурного анализа.
Опыт сравнительного анализа позволяет утверждать, что пространственная организация активного центра является наиболее консервативным элементом структуры гомологичных ферментов, в то время как остальные области могут принципиально отличаться [10]. Укладка полипептидной цепи и расположение боковых радикалов аминокислот в активном центре определяет характер взаимодействий, которые необходимы для узнавания, связывания и превращения субстрата. Кроме того, замечено, что аминокислотные остатки, влияющие на субстратную специфичность и каталитическую активность, как правило, находятся в радиусе 7-15Å от ключевых каталитических остатков [6]. В связи с этим сравнительный биоинформатический анализ наиболее важных с функциональной точки зрения элементов структуры – активных центров – представляет особый интерес.
Изучение молекулярных механизмов действия ферментов и использование этого знания для направленного дизайна их свойств остается важнейшей задачей современной биохимии. Разнообразие свойств в пределах одного семейства (субстратной специфичности, стереоселективности, стабильности и др.) является скорее правилом, чем исключением. В связи с этим актуальным представляется поиск и изучение таких специфических позиций подсемейств (СПП) в структурах белков одного семейства, изменчивость которых в процессе эволюции привела к разнообразию их функциональных характеристик – другими словами, аминокислотных позиций, которые консервативны только внутри определенных функциональных групп внутри целого семейства, но различаются между группами.
В работе использован новый алгоритм поиска специфических позиций подсемейств на основе анализа геномной и структурной информации. Проведен структурный сравнительный анализ ферментов суперсемейства альфа-бета гидролаз и рассмотрены каталитически важные позиции с точки зрения распределения типов аминокислот между ферментами с разными свойствами.
2. Методы
2.1 Поиск структур гомологичных белков. α/β-гидролазы с известной трехмерной структурой получали из базы данных ESTER. 95%-ный фильтр по аминокислотным последовательностям использовали для создания неоднородной выборки структур.
2.2 Идентификация структурных взаимосвязей в области активного центра. Базу данных структур активных центров готовили следующим образом. Для идентификации аминокислотных остатков активного центра, вовлеченных в каталитический механизм, использовали базу данных Catalytic Site Atlas (CSA). Аминокислотные остатки, обеспечивающие доставку, связывание и ориентацию субстрата в активном центре фермента, определяли с помощью алгоритма структурного анализа CASTp. Создавали координатный файл в формате PDB, содержащий область активного центра фермента (рис. 1). Фрагменты, состоящие менее чем из 6 аминокислот, удаляли. Остальные фрагменты, разделенные не более чем 10 аминокислотами, объединяли. Полученную библиотеку структурных файлов выравнивали с использованием программы Matt [5]. Для визуального структурного анализа использовали программу Pymol.
2.3 Оценка специфичности. Для того чтобы оценить степень специфичности каждой колонки множественного выравнивания, использовали следующую функцию оценки, основанную на формуле относительной энтропии: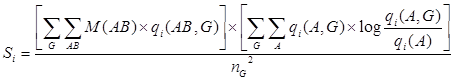
где A и B – типы аминокислот в колонке множественного выравнивания; qi(AB) и qi(AB, G) обозначают частоту пары AB в колонке i и в подсемействе G этой колонки; qi(A) и qi(A, G) – частоты аминокислоты типа А соответственно в колонке i и в подсемействе G; nG – общее количество подсемейств. M(AB) соответствует оценке биохимического сходства аминокислот типов A и B, рассчитанной как ![]() , где m(AB) – значение матрицы BLOSUM62. Диагональные элементы матрицы М(АА) приравнивали к 1 для того, чтобы не допустить переоценки редких типов аминокислот.
, где m(AB) – значение матрицы BLOSUM62. Диагональные элементы матрицы М(АА) приравнивали к 1 для того, чтобы не допустить переоценки редких типов аминокислот.
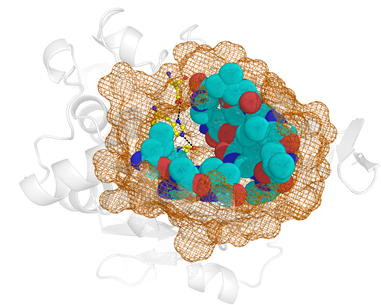
Рис 1. Область активного центра – фрагмент полноразмерной структуры фермента, состоящий из (1) остатков, обеспечивающих функционирование каталитического механизма (показаны в виде ball-and-sticks); (2) остатков, которые взаимодействуют с функциональными группами субстрата в процессе его доставки и ориентации в активном центре (показаны как сферы); и (3) ряда окрестных остатков, формирующих целостность выделенного фрагмента (показаны в виде сетки).
Чтобы распознать, является ли распределение аминокислот по колонке случайным или ассоциировано с разбиением на подсемейства, каждую колонку перетасовывали 10 000 раз. При этом границы подсемейств оставляли неизменными [4]. Для каждой случайной перетасовки вычисляли оценку Sirnd.
2.4 Оценка консервативности. Для оценки консервативности колонки множественного выравнивания использовали формулу Valdar и Thornton [9].
2.5 Статистический анализ. Z-оценку специфичности и консервативности для каждой колонки рассчитывали как 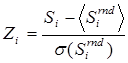 , где Si – оценка специфичности или консервативности колонки i, а Sirnd соответствует значению для случайно перетасованной колонки. Для расчета Р-оценок статистической значимости использовали ранговую статистику Бернулли [4].
, где Si – оценка специфичности или консервативности колонки i, а Sirnd соответствует значению для случайно перетасованной колонки. Для расчета Р-оценок статистической значимости использовали ранговую статистику Бернулли [4].
3. Результаты и обсуждение
Результатом дивергентной эволюции стало возникновение родственных ферментов, которые катализируют различные химические превращения, несмотря на структурное сходство активных центров. Понимание структурно-функциональных механизмов, которые обеспечивают различное функционирование гомологичных активных центров, представляет одну из важнейших задач современной энзимологии. Суперсемейство альфа-бета гидролаз является одной из наиболее крупных групп ферментов со схожей структурной организацией, но различными каталитическими свойствами [7]. Наблюдаемое разнообразие является следствием глубокой специализации от общего предка, которая сопровождалась потерей сходства по аминокислотным последовательностям. Например, представители суперсемейства липаза Б из Candida antarctica, сериновая карбоксипептидаза из Triticum aestivum и оксинитрилаза из Hevea brasiliensis характеризуются сходством по первичным структурам в пределах всего лишь 7,8–13,7%. Это не позволяет сравнивать удаленные гомологи при помощи выравнивания аминокислотных последовательностей, в то время как сравнение только близких гомологов не позволит охватить наблюдаемого в суперсемействе разнообразия свойств. Каноническое трехмерное выравнивание также не обнаруживает достаточного сходства пространственной организации указанных ферментов, что объясняется значительными различиями полноразмерных структур. Таким образом, сравнительный биоинформатический анализ эволюционно удаленных гомологов на уровне структурной организации активных центров (см. Методы, п. 2.2) был использован для изучения структурно-функциональных взаимосвязей в суперсемействе α/β-гидролаз. Полученное выравнивание 238 различных структур, по нашим данным, является самым масштабным множественным структурным сравнением белков с α/β-гидролазной укладкой полипептидной цепи на настоящий момент.
Сравнительный анализ показал, что очень небольшое количество участков имеют общую для всего суперсемейства структурную организацию, что можно объяснить глубокой специализацией от общего предка в процессе эволюции. 78 аминокислотных остатков структурно консервативны в 50% представителей суперсемейства, в то время как в 70% исследованных белков было обнаружено только 55 общих позиций, которые соответствуют основным структурным блокам α/β-гидролазной укладки [7] – β-слоям 3, 5, 6 и 7 центрального β-листа, а также α-спиралям C и E. Сравнительный анализ выявил значительное структурное сходство участков активного центра в области каталитической триады – петель между β5 и αC (так называемая петля нуклеофила), β7 и αE, а также β8 и αF. Статистический анализ множественного выравнивания показал, что только три аминокислотных остатка можно считать консервативными во всем суперсемействе: каталитический гистидин, а также два остатка глицина, формирующих консенсусный паттерн GXNXG, где N обозначает положение каталитического нуклеофила, а Х допускает присутствие любой аминокислоты (таблица 1). Показано, что упомянутые позиции не только содержат аминокислоты одного функционального типа, но также занимают одно положение и ориентацию в структурах ферментов суперсемейства. Тем не менее единственной строго консервативной аминокислотой является только каталитический гистидин. В то время как роль этого остатка в разных каталитических механизмах может варьировать, эволюционное давление на эту позицию, по всей видимости, говорит об уникальных химических свойствах гистидина в реакциях переноса протона.
Таблица 1 – Аминокислоты, консервативные в суперсемействе α/β-гидролаз – каталитический гистидин (His), а также глицины консенсусного паттерна нуклеофила Gly1XNXGly2
|
Ранг |
Z-оценка |
Р-оценка |
Позиция |
Аминокислотный состав |
|
1 |
4.89 |
2.72*10-5 |
His |
H (100%) |
|
2 |
4.23 |
4.33*10-11 |
Gly1 |
G (91%) A (4%) S (2%) L (1%) E (1%) W (<1%) H (<1%) |
|
3 |
4.10 |
6.03*10-14 |
Gly2 |
G (90%) A (4%) S (2%) V (1%) T (1%) I (1%) C (<1%) L (<1%) H (<1%) |
Таблица 2 – Специфические позиции подсемейств в суперсемействе α/β-гидролаз
|
Ранг |
Z |
Р |
Позиция |
П1 |
П2 |
П3 |
П4 |
|
1 |
49.0 |
0.0 |
Ser 105 |
C(100%) |
D(100%) |
S(100%) |
S(100%) |
|
2 |
17.6 |
1.4* 10-134 |
Asp 187 |
D(100%) |
D(100%) |
D(100%) |
E(100%) |
|
3 |
9.7 |
1.4* 10-62 |
Gln 106 |
W(67%) L(33%) |
W(75%) Y(13%) I(12%) |
M(20%) L(19%) Q(18%) Y(11%) A(10%) N(5%) F(4%) T(3%) W(2%) V(2%) S(1%) C(1%) K(1%) G(1%) D(1%) H(1%) |
A(65%) M(15%) L(8%) F(8%) G(4%) |
|
9 |
6.0 |
7.1* 10-71 |
Thr 40
|
I(67%) A(33%)
|
F(63%) W(13%) E(13%) gap(11%) |
G(18%) L(15%) F(12%) T (10%) gap(10%) S(7%) Y(6%) W(5%) I(4%) A(4%) M(2%) N(1%) E(1%) V(1%) R(1%) H (1%) D(1%) C(1%) |
G(65%) gap(15%) L(12%) A(4%) S(4%) |
Подсемейство 1 (П1) включает диенлактон-гидролазы, П2 – эпоксидгидролазы, П3 – пролилэндопептидазы, карбоксипептидазы, галопероксидазы, кутиназы, липазы и оксинитрилазы, П4 – некоторые карбоксиэстеразы, ацетилхолинэстеразы и бутирилхолинэстеразы. Нумерация позиций приведена в соответствии с липазой Б из Candida antarctica. Для каждой позиции указан ранг ее статистической значимости, Z- и P-оценки, встречаемость типов аминокислот в подсемействах.
Анализ множественного выравнивания двух других остатков каталитической триады – нуклеофила и аминокислоты, а также остатков оксианионного центра показал специфическое распределение типов аминокислот в этих позициях среди подсемейств ферментов с различными свойствами (таблица 2). Так, например, серин, аспартат и цистеин могут играть роль нуклеофила в ферментах из разных подсемейств, в то время как позиция каталитической кислоты может быть занята аспартатом или глутаматом. Описанное наблюдение может показаться тривиальным, однако является хорошей иллюстрацией того, как систематический анализ специфических позиций может быть использован для изучения разнообразия свойств и структурно-функциональных взаимосвязей в ферментах. Кроме того, сайт-направленный мутагенез каталитического нуклеофила (специфической позиции) часто используется в белковой инженерии для дизайна ферментов с новыми свойствами [3]. Другим интересным наблюдением является специфичность остатков оксианионного центра – гомологов Thr40 и Gln106 CALB в других ферментах. Как правило, только атомы основной цепи этих аминокислот участвуют в катализе, однако боковые радикалы могут образовывать стабилизирующие взаимодействия с «якорными» остатками [8]. Можно предположить, что аминокислотный состав этих СПП меняется в зависимости от окружения в ферментах из различных подсемейств, что подразумевает структурную роль.
4. Выводы
Использован алгоритм структурного анализа эволюционно родственных, но удаленных ферментов, претерпевших значительные изменения в ходе естественного отбора и специализации от общего предка. Биоинформатический анализ применен к суперсемейству α/β-гидролаз для изучения таких позиций в структурах белков, специфическая изменчивость которых позволяет предположить их ведущую роль в формировании функционального разнообразия. Сравнительный анализ ферментов суперсемейства показал высокую степень сходства основных структурных блоков α/β-гидролазной укладки – β-слоев 3, 5, 6 и 7 центрального β-листа, а также α-спиралей C и E. Показано, что каталитический гистидин является единственной строго консервативной аминокислотой среди α/β-гидролаз. Остальные аминокислоты каталитической триады, а также остатки оксианионного центра были определены как специфические позиции подсемейств. Распределение типов аминокислот в этих позициях соответствует разделению ферментов на подсемейства с различными каталитическими активностями. Данный алгоритм может быть использован также для систематического изучения структурно-функциональных взаимосвязей в семействах ферментов на основе биоинформатического анализа и определения специфических позиций. Информация о специфических позициях может быть использована для получения ферментов с заданными свойствами, а также функциональной аннотации недавно открытых белков.
Работа была поддержана Министерством науки и образования РФ (госконтракт 02.740.11.0866) и Европейской комиссией (соглашение 227279) в рамках совместного научного проекта Россия-ЕС FP7-KBBE-2008-2B «IRENE».
Рецензенты:
Муронец Владимир Израилевич, д.б.н., профессор, заведующий отделом биохимии животной клетки НИИ физико-химической биологии имени А.Н. Белозерского МГУ имени М.В. Ломоносова, г. Москва.
Долинная Нина Германовна, д.х.н., ведущий научный сотрудник кафедры химии природных соединений химического факультета МГУ имени М.В. Ломоносова, г. Москва.
Библиографическая ссылка
Шаповалова И.В., Панин Н.В., Тахавеев В.А., Мисюра Н.М., Суплатов Д.А. БИОИНФОРМАТИЧЕСКИЙ АНАЛИЗ СТРУКТУРНЫХ ВЗАИМОСВЯЗЕЙ В УДАЛЕННЫХ ГОМОЛОГАХ СУПЕРСЕМЕЙСТВА АЛЬФА-БЕТА ГИДРОЛАЗ // Современные проблемы науки и образования. 2012. № 6. ;URL: https://science-education.ru/ru/article/view?id=7529 (дата обращения: 27.06.2026).