



Рак толстой кишки (РТК) относится к наиболее распространенным онкологическим заболеваниям [1]. Для РТК характерны высокие темпы роста заболеваемости и смертности, сохраняющиеся на протяжении последних десятилетий. Ежегодное число заболевших РТК во всём мире близится к 1,5 миллионам человек. В 2012 году зарегистрировано 1 млн 360 тыс. человек. В России показатель распространенности злокачественными новообразованиями в 2016 г. составил 2 403,5 на 100 000 населения, что выше уровня 2006 г. (1 730,9) на 38,8 % [2]. В структуре заболеваемости злокачественными новообразованиями в России рак толстой кишки занимает 4-е место. В 2016 г. в РФ из взятых на учет 530509 больных с впервые установленным диагнозом злокачественные новообразования (ЗНО), 60275 пришлось на колоректальный рак, что составило 11,3 % [2]. С 2005 по 2015 г. в России абсолютный прирост заболеваемости раком толстой кишки составил 26,09 %. В структуре смертности населения РФ от злокачественных новообразований удельный вес рака ободочной кишки составляет 7,9 %, прямой кишки – 5,7 % [2].
В большинстве случаев рак толстой кишки носит спорадический характер. Генетические факторы оказываются значимыми в 15–30 % случаев. Однако только около 5 % всех форм колоректального рака развивается на фоне хорошо известных наследственных синдромов, таких как синдром Линча (наследственный неполипозный рак толстой кишки, ННПРТК), семейный аденоматозный полипоз (САП) и MUTYH-ассоциированный полипоз (МАП) [3].
Семейный аденоматозный полипоз (САП) – тяжелое аутосомно-доминантное заболевание, развивающееся в гене APC (AdenomatousPolyposisColi). Частота встречаемости данного синдрома колеблется от 1 на 6800 до 1 на 29000 человек [4]. Около 1 % от всех случаев рака толстой кишки связывают САП, который характеризуется преимущественным поражением толстой кишки множественными аденоматозными полипами и высоким индексом их малигнизации (до 100 %) [5]. Количество полипов толстой кишки может варьировать. Сотни и тысячи полипов характерны для классической формы заболевания, менее 100 для аттенуированной [6].
Мутации APC гена обнаруживаются в 80–90 % случаев классического САП и в 10-30 % аттенуированного САП (АСАП) [7]. Следует отметить, что олигополипоз может быть проявлением также и биаллельной мутации гена MUTYH [8]. АРС является геном-супрессором опухолевого роста. Он играет ключевую роль в работе wnt-сигнального пути, участвуя в деградации β-катенинав цитоплазме клеток. Мутации, изменяющие структуру белка APC, приводят к нарушению работы деградирующего комплекса (GSK3β, аксин-1 и APC) и увеличению концентрации β-катенина. В результате накапливающийся в цитоплазме свободный β-катенин проникает в ядро и активирует транскрипцию некоторых генов и онкогенов, контролирующих клеточный рост и деление [9].
АРС-ген расположен на длинном плече пятой хромосомы (5q21-22). Состоит из 8535 пар оснований, организованных в 15 кодирующих экзонов. Белок содержит 2843 аминокислотных остатка. Мутации в АРС впервые были описаны в 1991 году и сегодня известно более 600 вариантов. Наиболее частыми являются мутации, приводящие к синтезу укороченного бека АРС: это мутации со сдвигом рамки считывания (68 %), нонсенс мутации (30 %), крупные делеции (2 %). Горячие точки гена локализованы в 1309 и 1061 кодонах, частота мутаций в которых составляет 17 % и 11 % соответственно [10]. Примерно у 10–30 % заболевание развивается вследствие мутации denovo [11,12].
Целью настоящего исследования стало определение спектра мутаций гена АРС у пациентов, проживающих на Юге России и проходивших диагностику и лечение на базе ФГБУ «РНИОИ».
Материалы и методы
Поиск наследственных мутаций проводился у 7 пациентов с клиническими признаками семейного аденоматозного полипоза (САП) и у всех больных изучался семейный анамнез.
У пяти пациентов наблюдалась классическая форма САП, характеризующаяся тысячами полипов на протяжении всей толстой кишки, а у двоих – аттенуированная форма с менее чем 30 колоректальными аденомами. Выделение геномной ДНК было выполнено из лейкоцитов переферической крови по стандартной методике, используя фенол-хлороформную экстракцию. Концентрацию полученных препаратов ДНК измеряли на флюориметре «Qubit 2.0» (Invitrogen, USA) с помощью набора Quant-iTТМdsDNA и нормализовывали её до 2 нг/мкл. 15 кодирующих экзонов гена АРС с примыкающими частями интронов (50–100 пар нуклеотидов) амплифицировали методом полимеразной цепной реакции с использованием 23 пар праймеров. В случае аттенуированного полипоза также проводилось исследование 16 кодирующих экзонов гена MUTYH с использованием 9 пар праймеров. Далее полученные фрагменты ДНК секвенировали по двум комплементарным цепям с использованием ABI PRISM 3500 (8 capillaries; Applied Biosystems).
Результаты
Средний возраст постановки диагноза составил 31,3 года. Диагноз САП был подтвержден гистологически у всех пробандов. В исследуемой группе больных было выявлено пять пациентов с герминальной мутацией в гене АРС, что составило 71,4 %. Поиск мутаций был проведен у 12 кровных родственников пациентов. У 4 человек были выявлены аналогичные варианты. Все семьи прошли медико-генетическое консультирование с рекомендациями по осуществлению пожизненного клинического мониторинга лицам с выявленными мутациями (таблица).
Данные об исследованных пациентах с обнаруженной герминальной мутацией в гене АРС.
|
Пациент |
Возраст возникнове-ния полипоза |
Количество полипов |
Мутация |
Количество родственников с мутацией |
|
1 |
22 года |
Более 1000 полипов |
847C>T(p.Arg283Term) |
1 (сын) |
|
2 |
40 лет |
Более 1000 полипов |
c.2362A>T(p.Lys788Term) |
? |
|
3 |
28 лет |
Более 1000 полипов |
c.2365C>T(p.Gln789Term) |
0 |
|
4 |
36 лет |
Более 1000 полипов |
с.1744-2A>G |
1 (сын) |
|
5 |
33 года |
Более 1000 полипов |
1309del5(c.3927_3931delAAAGA) |
2 (брат и племянница) |
Четыре пациента имели родственника первой степени родства с аденоматозным полипозом, и у одного пациента с высокой вероятностью мутация возникла de novo. У пациентов с аттенуированной формой полипозамутации в генах APC и MUTYH обнаружены не были. Герминальные мутации были представлены следующими типами: три нонсенс-мутации (60 %), одна делеция (20 %) и одна мутация сайта сплайсинга (20 %): 847C>Tp.Arg283Term), c.2362A>T(p.Lys788Term), c.2365C>T(p.Gln789Term), с.1744-2A>G, 1309del5(c.3927_3931delAAAGA) (рис. 1). Все они приводят к возникновению укороченного белка и, следовательно, являются истинно патогенными. Спектр выявленных мутаций несколько отличается от обнаруженных вариантов мутаций у населения центральной России [13].
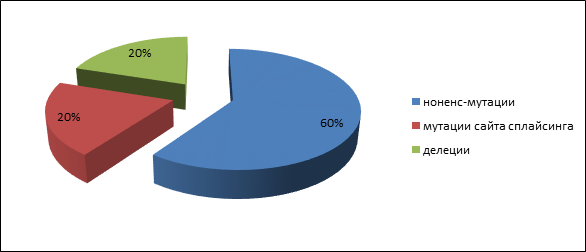
Рис. 1. Частота патогенных мутаций АРС гена
В качестве примера приводим клиническое описание и анализ родословной одного из пациентов с мутацией 1309del5(c.3927_3931delAAAGA)(рис. 2).
Пробанд К., 33 года, классическая форма САТК. При гистологическом исследовании выявлены тубулярные аденомы толстого кишечника с дисплазией 2 степени эпителия желёз. При колоноскопии обнаружено более тысячи полипов от 0,3 до 3 см во всех отделах толстой кишки. В семье отец Пробанда (II-4) в 36 лет прооперирован по поводу рака толстой кишки на фоне САТК, в 42 года умер. Братья и сестры отца здоровы. Бабушка Пробанда (I-1) умерла в 67 лет от рака почки, дедушка – в 72 года от рака легкого. Брат Пробанда (III-1) в 30 лет прооперирован в РОНЦ РАМН по поводу рака толстой кишки на фоне САТК. Проведено генетическое исследование детей Пробанда и брата. Мутация 1309del5 (рис. 3) выявлена у племянницы Пробанда, 7 мес. (IV-1).
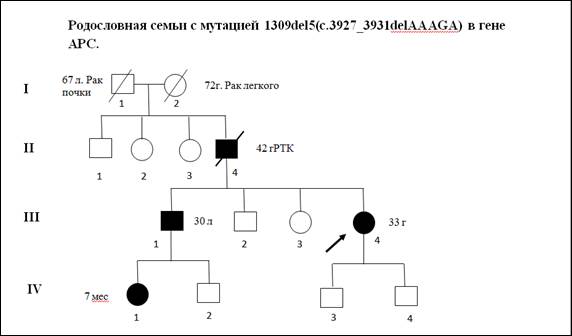
Рис. 2. Родословная семьи c семейным аденоматозным полипозом
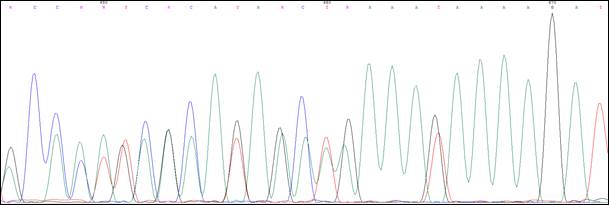
Рис. 3. Сиквенс пациента с мутацией 1309del5(c.3927_3931delАAAGA)
Обсуждение
Поиск мутаций при наследственных опухолевых синдромах позволяет определить тактику хирургического лечения, план последующей диспансеризации больного, а также помогает выявить родственников – носителей мутации. Это имеет важное значение при оценке риска развития злокачественных опухолей еще до клинической манифестации заболевания и способствует реализации стратегии персонализированной медицины и улучшению качества оказания онкологической медицинской помощи.Согласно рекомендациям американского общества по гастроэнтерологии (ACG), индивидуумы, имеющие более 10 колоректальных полипов, с наличием в семейном анамнезе родственника с синдромом полипоза, либо данные об аденомах и внекишечных проявлениях, характерных для САП (аденомы двенадцатиперстной кишки, десмоидные опухоли, папиллярный рак щитовидной железы, врожденная гипертрофия пигментного эпителия сетчатки, эпидермальные кисты, остеомы), должны быть обследованы на наличие синдромов аденоматозного полипоза. Генетическое тестирование включает анализ генов APC и MUTYH. Лицам, находящимся в группе риска по развитию САП, а также пациентам с уже имеющимися проявлениями САП с целью исключения малигнизации полипов необходимо ежегодное проведение колоноскопии или гибкой ректороманоскопии начиная с пубертата. В семьях с АСАП рекомендовано проведение колоноскопии. Абсолютными показаниями для неотложной колэктомии при САП, АСАП и МАП являются: подтвержденный или предполагаемый рак, либо наличие значимых симптомов злокачественного процесса. Относительные показания к операции включают наличие множественных аденом> 6 мм, увеличение числа аденом и невозможность адекватного обследования толстой кишки из-за множества мелких полипов. Эндоскопия верхних отделов ЖКТ показана пациентам в возрасте 25–30 лет и далее раз в 0,5–4 года в зависимости от стадии дуоденального полипоза по Шпигельману. 0 ст. – раз в 4 года, I ст. – раз в 2-3 года, II ст. – раз в 1-3 года, III ст. – раз в 6–12 мес, IVст.– хирургическое лечение. Также показано ежегодное УЗИ щитовидной железы. Детям до 7 лет раз в два года следует проводить исследование α-фетопротеина и УЗИ. Послеоперационное наблюдение за пациентами должно включать в себя ежегодную эндоскопию прямой кишки или кармана подвздошной кишки и осмотр илеостомы каждые два года [14]. Согласно рекомендациям Европейского Общества Медицинской Онкологии (ESMO)эндоскопическое обследование при классическом аденоматозном полипозе должно проводиться пожизненно. При бессимптомном носительстве мутации рекомендуется выполнение ректороманоскопии гибким фиброскопом каждые 2 года, начиная с 10–12-летнего возраста. При обнаружении хотя бы одной аденомы колоноскопию в последующем следует проводить ежегодно. Хирургическое лечение показано при большом количестве аденом, а также при наличии аденом с высокой степенью дисплазии. Эндоскопия верхних отделов ЖКТ проводится с 20–25 лет, либо с момента обнаружения колоректальных аденом. Частота проведения эндоскопии зависит от стадии дуоденального полипоза по Шпигельману. С целью исключения рака щитовидной железы показано ежегодное УЗИ. Диагностика десмоидных опухолей проводится с помощью КТ либо МРТ и может быть показана пациентам с семейным анамнезом, а также при локализации мутации в определенных сайтах гена АРС [15].
Заключение
Частота возникновения наследственных мутаций в гене APC у пациентов с классической формой САТК составила 71,4 %: три нонсенс-мутации (60 %), одна делеция (20 %) и одна мутация сайта сплайсинга (20 %). Полученные результаты указывают на необходимость исследования всех кодирующих экзонов гена APC у больных классической (тяжелой) формой семейного аденоматоза толстой кишки, а также обязательное обследование всех кровных родственников больного. При обнаружении мутации можно говорить о чрезвычайно высокой вероятности развития опухолевого синдрома (не менее 80 %) в течение жизни.