



Нейрофиброматоз представляет собой генетически гетерогенную группу наследственных моногенных заболеваний. В настоящее время описано 7 типов, из которых наибольшее клиническое значение имеют первые два.
Болезнь Реклингаузена (БР, нейрофиброматоз I типа, периферический нейрофиброматоз, НФ 1) относится к группе системных наследственных нейрокутанных факоматозов и характеризуется развитием опухолей эктодермального происхождения. Заболевание было описано в конце XIX века учеником Рудольфа Вирхова Фридрихом фон Реклингхаузеном. БР встречается с частотой 1:2000 – 1:3500 населения, в России 1,28:10000 [6; 7; 16; 20]. Оба пола болеют одинаково часто. Тип наследования аутосомно-доминантный с высокой пенетрантностью (близкой к 100%) и вариабельной экспрессивностью [5; 6; 20].
Ген НФ 1 был выявлен в 17 хромосоме на локусе q11.2 в 1987 году. Он экспрессируется в шванновских клетках, меланоцитах, лейкоцитах, клетках надпочечников, центральной нервной системы и кодирует белок нейрофибромин [7; 8; 16]. Белок содержит особый домен (НФ1-ГРД), который оказывает тормозное влияние на продукт проонкогена RAS и в норме ингибирует его функцию, за счет чего обеспечивается супрессорный эффект в отношении клеточной пролиферации. Воздействие проонкогена RAS не ограничивается участием в пролиферативных процессах, описано его влияние на формирование когнитивных нарушений в виде затруднения в процессе обучения чтению, письму, математике [6; 8; 18; 20].
Основными диагностическими критериями являются клинические симптомы, рекомендованные Международным комитетом экспертов по нейрофиброматозу, принятые в 1988 году. В соответствии с данными критериями диагноз БР может быть установлен при наличии у больного минимум 2 признаков [3; 4; 22].
1. Наличие 5 и более пигментных пятен цвета «кофе с молоком» диаметром более 5 мм у детей допубертатного возраста и не менее 6 пятен диаметром более 15 мм в постпубертатном возрасте.
2. Веснушчатость в подмышечных и/или паховых складках.
3. Не менее 2 нейрофибром любого типа или одна плексиформная нейрофиброма.
4. Дисплазия крыла клиновидной кости или врожденное истончение кортикального слоя длинных костей с псевдоартрозом или без него.
5. Глиома зрительного нерва.
6. Не менее 2 узелков Лиша (гамартомы) на радужке, выявляемые при исследовании с помощью щелевой лампы.
7. Наличие НФ 1 у родственников первой степени родства.
Из всех диагностических критериев кожные проявления являются наиболее значимыми, так как доступны при осмотре и зачастую бывают первыми симптомами болезни.
Пигментные пятна цвета «кофе с молоком» (франц. «café-at-lait») - первый и постоянный признак болезни Реклингаузена, встречающийся в 95% случаев. Пигментные пятна могут выявляться при рождении, но чаще появляются к трехлетнему возрасту на закрытых участках тела [3; 9; 13]. Они представляют собой монохромные пятна от светло-бежевого до тёмно-коричневого цвета, однородные по структуре, округлых или овальных очертаний, с ровными краями и четкими границами. Размеры могут варьировать от нескольких миллиметров до нескольких сантиметров, однако диагностически значимыми являются пятна диаметром более 5 мм у детей до пубертата и более 15 мм после полового созревания (рис. 1). Размер пятен увеличивается пропорционально росту ребенка [7; 16; 20]. При гистологическом исследовании в очагах обнаруживается повышенное содержание меланина в меланоцитах [2; 9; 13]. Пигментные пятна цвета «кофе с молоком» следует дифференцировать с гиперпигментированными невусами, меланоцитарными невусами, поствоспалительной гиперпигментацией.
Веснушки (симптом Кроува) – это пигментированные пятна размером 1-3 мм, светло-коричневого цвета, локализующиеся в подмышечной и/или паховой области, под молочными железами, как правило, появляющиеся на втором году жизни, реже в первые месяцы после рождения. Солнечное излучение не является провоцирующим фактором их возникновения, в отличие от обычных веснушек. Для их возникновения имеет значение трение и мацерация [2; 3; 5]. Частота выявления веснушек при болезни Реклингаузена варьирует от 21 до 93,7% [9; 13; 16].
Нейрофибромы – это доброкачественные опухоли, производные нервной оболочки периферических нервов. Они состоят из различных типов клеток: шванновских клеток, фибробластов, тучных клеток, эндотелиальных клеток, а также коллагеновых волокон [3; 10; 16]. Нейрофибромы представляют собой округлые узелки на коже и/или в толще кожи, мягкоэластической консистенции, синюшно-красного цвета и/ или цвета нормальной кожи, размерами от просяного зерна до 5 см и более (рис. 1, 2). При пальпации безболезненные, за исключением случаев вовлечения периферических нервов, когда возникают гипостезии и боли, иррадиирующие в соответствующие зоны иннервации [6; 7; 11].
В настоящее время нет единой классификации нейрофибром. M. Ruggieri и соавт. предлагают выделять поверхностные и глубокие образования, а J. Zeller и соавт. подразделяют их на кожные, подкожные и плексиформные [26; 29]. Такое различие происходит вследствие несоответствия клинических проявлений гистологическому расположению нейрофибром. A. Hernández-Martína и соавт. в 2016 году попытались объединить данные классификации. Авторы разделяют нейрофибромы на поверхностные и глубокие, при этом к поверхностным относят кожные (дермальные) и подкожные нейрофибромы, которые расположены в гиподерме, однако также могут иметь дермальный компонент. К глубоким относят плексиформную нейрофиброму [16].
Провоцирующим фактором роста нейрофибром является гормональная перестройка организма: пубертат, беременность. На начальном этапе развития кожные нейрофибромы могут не визуализироваться, в то же время с годами достигать больших размеров, куполообразно возвышаясь над поверхностью кожи. Таким образом, первые видимые элементы появляются в период полового созревания [5; 6; 11].
Нейрофибромы пальпируются как подкожные образования, расположенные линейно по ходу периферических нервов, мягкоэластической консистенции, при пальпации сдвигаются в поперечном направлении вместе с нервным стволом, характерным симптомом является проваливание пальца при легком надавливании (феномен «кнопка звонка»). Могут локализоваться на любом участке тела, однако несколько чаще встречаются на голове и шее, в этом случае их необходимо дифференцировать с увеличенными лимфоузлами [5; 7; 10; 16].
Глубокие плексиформные нейрофибромы обычно присутствуют с рождения и остаются незамеченными при клиническом обследовании до тех пор, пока не появляется неврологическая симптоматика (парестезии, парезы, потеря чувствительности, боль), или же они обнаруживаются при проведении МРТ-исследования [3; 16]. По данным U. Dagalakis и соавт., распространенность плексиформных нейрофибром у детей составляет 10%. У 8–13% больных БР они могут озлокачествляться, перерождаясь в нейрофибросаркому. О злокачественной трансформации свидетельствует непроходящая боль, быстрое увеличение размеров, появление уплотнений, неврологическая симптоматика [3; 12; 19].
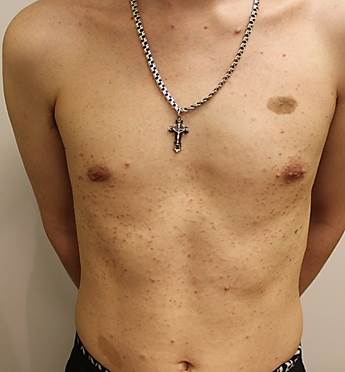
Рис. 1. Пациент А. 18 лет. Болезнь Реклингаузена. Пигментные пятна «кофе с молоком», множественные кожные нейрофибромы на коже передней брюшной стенки
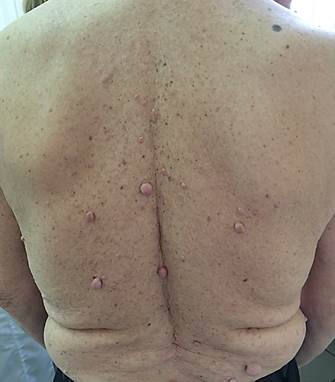
Рис. 2. Пациент В. 47 лет. Множественные кожные нейрофибромы на спине больного нейрофиброматозом I типа
Пятна «кофе с молоком» встречаются не только при БР, но и при других наследственных синдромах и заболеваниях. Дифференциальная диагностика проводится с нейрофиброматозом 2 типа, семейными пятнами café-at-lait, синдромом LEOPARD, синдромом McCune-Albright, синдромом Noonan и другими мультисистемными заболеваниями.
В частности, при нейрофиброматозе 2 типа пигментные пятна цвета «кофе с молоком» встречаются у 80% больных, однако диагностическая ценность их менее значима, чем при БР. Также более характерным является развитие шванном, чем нейрофибром. Из опухолей ЦНС преобладают менингиомы, глиомы, шванномы. Характерна ювенильная задняя субкапсулярная катаракта [5; 6; 16].
Семейный характер пятен café-at-lait должен быть заподозрен при выявлении у родственников разных поколений при отсутствии других проявлений нейрофиброматоза 1 типа. Тип наследования аутосомно-доминантный, однако до сих пор не описаны генетические мутации, приводящие к появлению пигментации [9].
Пигментные пятна у больных с синдромом LEOPARD, как правило, имеют более темную полихромную окраску, полигональные очертания. Кроме кожных проявлений, у пациентов обнаруживаются изменения ЭКГ, глазной гипертелоризм, стеноз легочной артерии, аномалии развития половых органов, нейросенсорная глухота, задержка роста и развития [1; 16; 23].
Для синдрома McCune-Albright характерно наличие пятен «кофе с молоком» бо?льших размеров, чем при БР, с неровными краями, нечеткими границами, полигональных очертаний, часто расположенных с одной стороны, сегментарно. Также у больных выявляется фиброзная дисплазия и гиперфункция эндокринных желез [9; 28].
Пациенты с синдромом Noonan фенотипически похожи на пациентов с нейрофиброматозом 1 типа. Больные, как правило, низкого роста с короткой шеей и треугольным лицом, для них характерно наличие пятен «кофе с молоком», веснушчатость в подмышечных и паховых областях, микроцефалия, птоз, стеноз легочной артерии, снижение интеллекта. Однако при синдроме Noonan не обнаруживаются кожные нейрофибромы, опухоли центральной нервной системы, узелки Лиша [3; 25].
Следует отметить, что характерные клинические признаки БР появляются у больных в разные периоды жизни, и по отдельности не являются патогномоничными, поэтому постановка окончательного диагноза может быть отсрочена на многие годы. А. Hernández-Martína и A. Duat-Rodríguezb в 2016 году сообщают, что в дополнение к классическим проявлениям БР (пятна «кофе с молоком», веснушки и нейрофибромы) у больных часто выявляются другие кожные симптомы, которые могут иметь прогностическую ценность у пациентов с неустановленным диагнозом. К ним относятся: анемический невус, ювенильная ксантогранулема, диффузная гиперпигментация, гипопигментированные пятна [17].
Наиболее часто выявляется анемический невус. Впервые связь между болезнью Реклингаузена и анемическим невусом предположил Naegeli в 1915 г. Анемический невус представляет собой бледное неправильных очертаний пятно размерами от нескольких миллиметров до нескольких сантиметров. Невус может быть одиночным или множественным и располагаться на любом участке, чаще встречается в окологрудинной области. Клинически он не всегда обнаруживается и проявляется после незначительного трения кожных покровов, при этом оставаясь неизменным бледным пятном на фоне общей гиперемии здоровой кожи [11; 17; 27]. M. Marque и соавт. отмечают высокую распространенность анемических невусов у детей, однако зачастую они обнаруживаются в более позднем возрасте при осмотре дерматологом, так как остаются незаметными для родителей. Впоследствии у 50% детей с анемическими невусами был подтвержден диагноз «болезнь Реклингаузена» [17; 21].
Ювенильная ксантогранулема является наиболее распространенной формой гистиоцитоза. Она представляет собой папулу округлой или овальной формы, плотноватой консистенции, размерами от 1 до 2 см, желтоватого или красно-желтого цвета, безболезненная при пальпации. Характерна спонтанная инволюция в пубертатном периоде. Ювенильные ксантогранулемы могут быть единичными и множественными. Локализуются чаще всего на лице, волосистой части головы, туловище, реже на слизистых оболочках. По данным M. Fenot (2014), распространенность ювенильной ксантогранулемы у больных БР варьирует от 0,7% у взрослых до 37,5% у детей, особенно у детей в возрасте до 9 лет [14; 15; 24].
Также у пациентов с болезнью Реклингаузена может встречаться зуд, диффузная гипер- и/ или гипопигментация. Зуд беспокоит 20% больных БР и существенно снижает их качество жизни. Диффузная гиперпигментация отчетливо выявляется при сравнении тона кожи больного с тоном кожи других здоровых членов семьи [7; 17].
Нейрофиброматоз опасен прежде всего своими осложнениями, возникающими при несвоевременной диагностике, что обуславливает необходимость диспансерного наблюдения. Наиболее прогностически неблагоприятным осложнением БР является озлокачествление имеющихся нейрофибром. Также к осложнениям НФ 1 относятся глиома зрительных нервов, приводящая к слепоте, феохромоцитома с развитием симптоматической злокачественной артериальной гипертензии. При БР описаны васкулиты, приводящие к стенозу почечных артерий, коарктации аорты [6; 10; 11].
Таким образом, ранняя диагностика и диспансеризация больных нейрофиброматозом имеет решающее значение в прогнозе и качестве жизни пациентов. Повышение знаний об указанной патологии у специалистов разного профиля будет способствовать своевременной диагностике НФ 1 и выявлению осложнений. Болезнь Реклингаузена является одним из заболеваний, где важна преемственность в работе врачей разных специальностей.
Библиографическая ссылка
Уфимцева М.А., Бочкарев Ю.М., Гальперин А.М., Головырина И.Л., Гурковская Е.П. КОЖНЫЕ ПРОЯВЛЕНИЯ БОЛЕЗНИ РЕКЛИНГАУЗЕНА // Современные проблемы науки и образования. – 2016. – № 6. ;URL: https://science-education.ru/ru/article/view?id=25818 (дата обращения: 19.04.2024).