



Болезнь Альцгеймера: актуальность проблемы и патогенез
Болезнь Альцгеймера (БА) является нейродегенеративной патологией, развивающейся преимущественно у людей пожилого и старческого возраста. Наблюдаемое в последнее десятилетие неуклонное старение населения в развитых странах мира приводит к увеличению частоты возникновения нейродегенеративных заболеваний. В современном мире эта патология занимает 6-е место по причине смертности. По оценкам Всемирной организации здравоохранения, статистика частоты встречаемости БА в ближайшем будущем неутешительна. По разным данным, БА диагностирована у 14–26 миллионов человек, ежегодно выявляется 4,6 миллиона новых случаев деменции. В Российской Федерации БА страдают 1,4 миллиона человек. По прогнозам распространенность деменции в развитых странах с 2001 по 2040 гг. увеличится на 100%, а в развивающихся странах (Индия, Китай, страны Юго-Восточной Азии) – более чем на 300%. Современные исследователи выделяют еще один аспект медико-социальной проблемы БА – экономический. В связи с увеличением доли пожилых людей современное общество вынуждено все больше средств тратить на дорогостоящее лечение пациентов с БА. В связи с этим БА представляет собой актуальную медико-социальную проблему.
Патогенез БА связан с поражением гиппокампа и других областей головного мозга, ответственных за формирование памяти [1, 2]. Основным гистологическим признаком БА является накопление в головном мозге пациентов сенильных бляшек и нейрофибриллярных клубков. Формирование нейрофибриллярных клубков в основном связано с внутриклеточным накоплением в нейронах гиперфосфорилированного τ-протеина, в то время как сенильные бляшки являются внеклеточными отложениями агрегатов различных бета-амилоидных пептидов (Aβ). Главенствующей теорией возникновения БА на сегодняшний день является «амилоидная гипотеза», согласно которой повышенная продукция пептида Aβ длиной в 42 аминокислотных остатка (Aβ42) приводит к потере синаптических связей нейронов в гиппокампе, коре и субкортикальных областях головного мозга [2, 3]. Тау-гипотеза патогенеза БА предполагает, что нарушения в метаболизме тау-белка, ассоциированного с микротрубочками, приводят к его скоплению и агрегации, в результате чего нарушается аксональный транспорт и развиваются нейродегенеративные изменения центральной нервной системы [4]. Кальциевая гипотеза утверждает, что первопричиной развития БА является нарушение кальциевого сигналинга в нейронах, приводящее к накоплению кальция в цитоплазме и повышенной уязвимости нейронов к эксайтотоксичности [5]. Известны гены, ассоциированные с БА, такие как APP, PSEN, ApoE [6]. Однако ни «амилоидная гипотеза», ни другие теории происхождения БА не дают полного представления о причине этого заболевания и его нейроиммуноэндокринных механизмах [5]. Новой является гипотеза о роли «inflamm-aging» в патогенезе нейродегенеративных заболеваний, в том числе БА. В связи с этим целью обзора явилась систематизация данных о предполагаемой роли нарушений функций иммунной системы при ее старении в патогенезе БА.
Предполагаемая роль хронического воспаления (inflamm-aging) в патогенезе болезни Альцгеймера
Inflamm-aging – термин, описывающий слабовыраженное хроническое воспаление, возникающее при старении организма при отсутствии явного инфекционного поражения [7, 8]. Предположительно, такая слабая хроническая воспалительная реакция может возникать при ускоренном старении иммунной системы в сочетании с окислительным стрессом, вызываемым дисфункцией митохондрий (рис. 1), и проводить к развитию ряда заболеваний [9] (рис. 2).
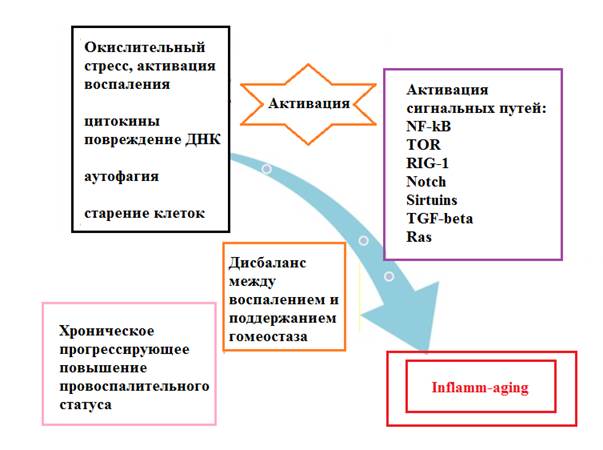
Рис. 1. Схематическое изображение развития слабой хронической воспалительной реакции при ускоренном старении иммунной системы в сочетании с окислительным стрессом, вызываемым дисфункцией митохондрий. Адаптировано из S. Xia et al. [9]
Об inflamm-aging как о системном воспалительном ответе свидетельствует повышенный уровень провоспалительных цитокинов и факторов свертываемости в сыворотке крови при старении [10–12].
Как известно, при БА ранним проявлением патологии является потеря синапсов, в результате которой происходят дальнейшие нарушения синаптической передачи и ухудшение когнитивных функций. Существует мнение, что потеря синапсов при БА опосредована системой комплемента.
Система комплемента может быть активирована тремя различными путями. Классический путь обычно инициируется комплексом «антиген–антитело», образование которого приводит к фагоцитозу и/или возникновению пор в мембране, лизису и гибели клеток. Альтернативный путь постоянно активен, не требует образования иммунных комплексов и срабатывает сразу же после появления антигенов. Лектиновый путь активируется маннан-связывающим лектином [13]. Синаптическая потеря, опосредованная системой комплемента, вероятно, происходит по классическому пути. Этот путь инициируется, когда комплекс «антиген–антитело» связывается с белком C1q, хотя существуют работы, в которых описана независимая активация C1q [14, 15].
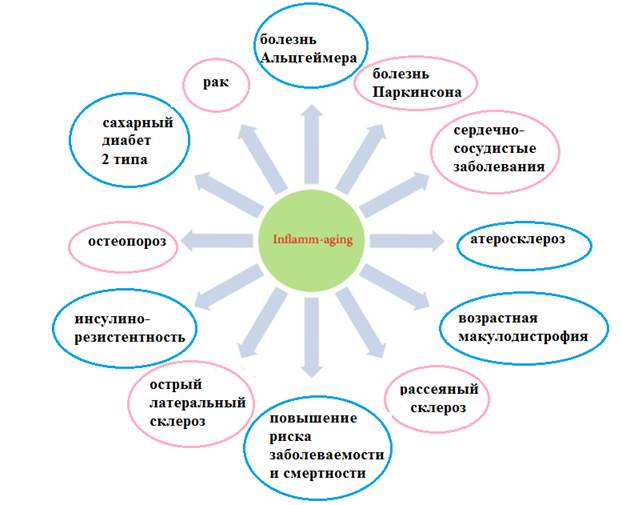
Рис. 2. Роль inflamm-aging в патогенезе различных заболеваний.
(адаптировано из S. Xia et al. [9])
В работе S. Hong и соавт. (16) продемонстрировано, что при БА Aβ и гиперфосфорилированный τ-протеин активируют путь комплемента, связываясь с компонентом C1q, что приводит к активации классического пути комплемента и потере синапсов. По мнению авторов, ключевую роль в этом процессе играет микроглия [16]. Микроглия занимает важное место в фагоцитозе амилоидного белка и в воспалительной реакции при БА. Выделены два механизма, по которым происходит утилизация токсичных амилоидных олигомеров в головном мозге. Первый опосредуется несколькими рецепторами, которые экспрессируются в микроглии, в том числе через скавенджер рецепторы, рецепторы системы комплемента – FcRs и TREM2. Второй путь инициирует деградацию амилоидов при участии ферментов: неприлизина, инсулин-разрушающих ферментов, матриксных металлопротеаз и катепсина B. Опосредованное микроглией очищение мозга от амилоидов зависит от возраста и стадии развития БА. Фагоцитарную активность микроглии ослабляет провоспалительный эффект цитокинов – интерферона-γ (INF-γ), интерлейкина-1 (IL-1), фактора некроза опухолей-α (TNF-a), которые, вероятно, склоняют активность микроглии в сторону провоспалительного фенотипа. В другом исследовании продемонстрировали локализацию C3 компонента комплемента на реактивных астроцитах при БА у человека [17], что также может способствовать потере синапсов.
Возможный механизм элиминации синапсов при БА, опосредованный системой комплемента, представлен на рисунке 3. БА характеризуется прогрессирующим накоплением в тканях головного мозга внеклеточного и внутриклеточного пептида Aβ, глиозом и нейровоспалением. Нейрональный C1q и микроглиальный C1q рекрутируются в синапсы и взаимодействуют с пептидом Aβ. Это запускает активацию белка С3 комплемента, экспрессируемого астроцитами и микроглией. C3 расщепляется на фрагменты C3b и iC3b, которые «маркируют» синапсы и связываются с CR3 в микроглии. Все эти события приводят к удалению «маркированных» синапсов [18].
Иммунологическая гипотеза БА подтверждается множеством исследований, посвященных изучению цитокинового профиля пациентов с БА. При этом, как и в случае с системой комплемента, считается, что высвобождение медиаторов воспаления происходит вследствие активации микроглии и астроцитов цитотоксическим окружением сенильных бляшек и нейрофибриллярных клубков [19]. Гипотезу о роли нейровоспаления в патогенезе БА косвенно подтверждает тот факт, что у лиц, принимающих нестероидные противовоспалительные препараты, риск развития БА снижен [20].
R. Taipa и соавт. (21) изучали взаимосвязь про- и противовоспалительных цитокинов в спинномозговой жидкости (СМЖ) и уровня когнитивных нарушений у пациентов с БА. Авторы определяли уровни 27 цитокинов в СМЖ пациентов с БА и лиц без патологии центральной нервной системы. Кроме того, исследовали корреляцию уровня цитокинов с когнитивным статусом и прогрессированием заболевания через 12 месяцев. Продемонстрировано, что пациенты с БА имели более высокие уровни провоспалительных и противовоспалительных цитокинов (эотаксин, IL-1, IL-4, IL-7, IL-8, IL-9, IL-10, IL-15, гранулоцитарно-макрофагальный колониестимулирующий фактор, моноцитарный хемоаттрактантный протеин-1, тромбоцитарный фактор роста, фактор некроза опухоли-α) по сравнению с контролем [21].
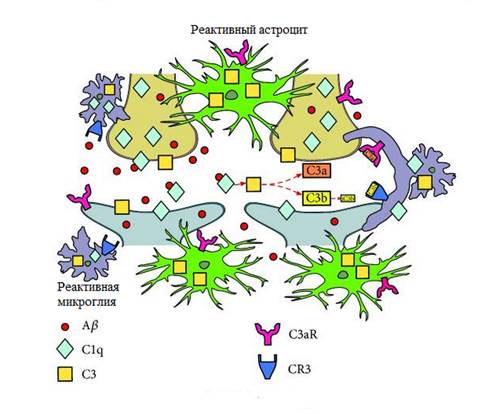
Рис. 3. Возможный механизм элиминации синапсов при БА, опосредованный системой комплемента, адаптировано из С. Luchena et al. [18], пояснения в тексте
Наблюдалась отрицательная корреляция между прогрессированием заболевания и уровнями некоторых цитокинов (IL-1β, IL-4, IL-6, IL-9, IL-17A, основной фактор роста фибробластов, гранулоцитарно-макрофагальный колониестимулирующий фактор, интерферон-γ, макрофагальный белок воспаления-1β). Авторы делают вывод о «защитной» роли повышения уровня специфических интратекальных цитокинов при БА и предполагают, что «перебалансировка» иммунной системы представляет новую цель в терапевтическом подходе к БА [22]. Влияние цитокинов на когнитивный статус пациентов также было продемонстрировано ранее в нескольких исследованиях [23, 24].
По некоторым данным, определенные полиморфизмы генов TNF-a, IL-1, IL-6, IL-10 увеличивают риск развития БА [25, 26, 27]. В пользу гипотезы о нейровоспалении свидетельствуют данные о том, что иммунотерапия снижает риск развития БА. Работа G. Mudò и соавт. (28) посвящена изучению IFNβ1a и его противовоспалительных эффектов в модели БА у крыс. Подкожное введение IFNβ1a в течение двух недель снижало выраженность когнитивных нарушений и предотвращало активацию микроглии и секрецию провоспалительных цитокинов (IL-6, IL-1β) в гиппокампе в модели БА у животных. Уровень экспрессии IL-10, значительно сниженный у животных с моделью БА, восстанавливался до контрольных значений после лечения IFNβ1a [28].
Перспективы применения периферических тканей для прижизненной диагностики болезни Альцгеймера
Известно, что развитие БА сопровождается изменением синтеза сигнальных молекул (пептида Аβ42, τ-протеина, α-синуклеина и др.) не только в головном мозге, но и в других тканях – в лимфоцитах крови и СМЖ. Клетки буккального эпителия легко доступны (соскоб со слизистой поверхности щеки) и при использовании метода иммуноцитохимии могут указывать на ускоренное старение организма или возникновение различной соматической патологии. Ранее полученные нами данные демонстрируют увеличение экспрессии маркеров клеточного старения p16 и p53 в клетках гиппокампа, буккального эпителия, фибробластов кожи при БА [29]. Имеющиеся литературные данные свидетельствуют о повышении экспрессии маркера клеточного старения p53 при БА в тканях головного мозга. Полученные ранее данные об изменении интенсивности экспрессии ассоциированных с БА и БП сигнальных молекул (пептид Аβ42, τ-протеин, протеинкиназа С, α-синуклеин, маркеры клеточного старения и апоптоза р16 и р53) в буккальном эпителии и фибробластах кожи позволяют сделать вывод о целесообразности использования указанных периферических тканей для ранней диагностики БА [30]. В лимфоцитах и тромбоцитах крови при БА выявлены увеличение количества τ-протеина, уменьшение длины теломер и изменение уровня других молекулярных маркеров по сравнению с лицами без патологии ЦНС [29].
Заключение. Таким образом, важную роль в патогенезе БА играют нейроиммунные взаимодействия, в результате которых развивается воспаление, приводящее к гибели нейронов и потере синапсов. Установлено, что нейроиммунные взаимодействия в тканях головного мозга опосредованы про- и противовоспалительными цитокинами, а также системой комплемента. Это позволяет предположить, что для диагностики БА важную роль могут играть не только ткани головного мозга, недоступные при жизни пациента, но и различные периферические ткани, участвующие в нейроиммуноэндокринной регуляции.
Дальнейшее развитие иммунологической теории патогенеза БА даст возможность модернизировать существующую диагностику БА (например, при помощи оценки цитокинового профиля пациента), что позволит разработать критерии для иммунотерапии этого нейродегенеративного заболевания.
Библиографическая ссылка
Зуев В.А. ИММУНОЛОГИЧЕСКАЯ ТЕОРИЯ ПАТОГЕНЕЗА БОЛЕЗНИ АЛЬЦГЕЙМЕРА: ФАКТЫ И ГИПОТЕЗЫ // Современные проблемы науки и образования. – 2019. – № 4. ;URL: https://science-education.ru/ru/article/view?id=28961 (дата обращения: 24.04.2024).