



В настоящее время болезнь Альцгеймера (БА) является наиболее распространенным, длительно протекающим нейродегенеративным заболеванием. На начальном этапе болезнь проявляется нарастающим ухудшением кратковременной памяти. Позднее наблюдаются поведенческие нарушения, происходит потеря долговременной памяти, затем следует деградация систем жизнеобеспечения организма, что в конечном итоге приводит к смерти [1]. Отложение амилоида β (Aβ) во внеклеточном матриксе головного мозга (ГМ) является типовым патологическим признаком при БА. В основном это Аβ40 и Aβ42 (по количеству аминокислот), причем Aβ42 более склонен к агрегации и более токсичен для клеток мозга [2]. Токсичность Aβ объясняется его биохимическими свойствами, способствующими дисмеризму в так называемые бляшки, воздействующие на нейроны и клетки глии. По данным исследований, амилоидные бляшки вызывают воспалительный ответ [3] и сопутствующую дисфункцию митохондрий [4]. Снижается синаптическая пластичность, происходят сбои во внутриклеточных сигнальных каскадах, в результате нейроны деградируют [5]. Бляшки – не единственный признак, внутри нейронов при БА наблюдаются нейрофибриллярные клубки (NFT) – скопления нитей гиперфосфорилированного Тау-белка [6]. В норме этот белок связан с тубулином микротрубочек и стабилизирует их структуру. Гиперфосфорилированнный Тау диссоциирует от микротрубочек, вызывая коллапс микротрубочкового цитоскелета, в результате нарушаются аксональный транспорт и синаптический метаболизм [7]. Исследования, направленные на ингибирование гликогенсинтетазы киназы-3β (GSK3β), фермента, регулирующего фосфорилирование Тау-белка (сверхактивная GSK3β приводит к гиперфосфорилированию Тау), предотвращают деградацию нейронов [8]. Чрезмерная активность GSK3β на ранних стадиях БА может вызывать перепроизводство Aβ и в дальнейшем гиперфосфориляцию Тау-белка [9]. Однако, несмотря на весомые аргументы за первостепенную роль Aβ, в исследованиях не была обнаружена устойчивая корреляция между плотностью и количеством бляшек в мозге и тяжестью деменции [10]. Корреляция прослеживается с плотностью клубков гиперфосфорилированного Тау, но не объясняет все патологические изменения [10]. Основным же коррелятом ухудшений при БА является потеря синапсов и холинергических нейронов, что и вызывает когнитивные нарушения [11, 12]. В холинергических нейронах снижено производство холин-ацетилтрансферазы (СhAT), катализирующей реакцию образования нейромедиатора ацетилхолина [13]. Ингибиторы ацетилхолинэстеразы (AChE), фермента, катализирующего гидролиз ацетилхолина до холина и уксусной кислоты, повышают уровень ацетилхолина в синаптической щели и частично улучшают состояние пациентов, но на очень непродолжительный период, не прекращая дальнейшую дегенерацию [14]. Несмотря на многочисленные исследования, в основном ориентированные на нейронные процессы, на сегодняшний день не разработано ни одного препарата, позволяющего вылечить болезнь. Комплекс профилактических мер, включающий в себя диету, физическую активность и умственные нагрузки, показывает некоторую эффективность в борьбе с заболеванием [15].
Заболевание классифицируют на раннюю и позднюю формы. Поздняя (спорадическая) форма наиболее распространена (более 90% случаев) среди людей старше 65 лет [1]. Основным генетическим фактором риска для позднего начала заболевания является ген APOE в хромосоме 19 [1, 16]. Генетически наследуемая форма БА с ранним началом (до 60–65 лет) может быть вызвана следующими мутациями: в гене APP, который кодирует белок-предшественник амилоида (БПА), в гене пресенелина 1 (PSEN1) в хромосоме 14 и/или в гене пресенелина 2 (PSEN2) в хромосоме 1 [1, 16]. Мутации в пресенелинах приводят к изменению обработки БПА и, кроме того, к нарушению передачи Ca2+-сигналов. Так, мутации PSEN1 приводят к увеличению выделения Ca2+ из эндоплазматического ретикулума (ЭР) с помощью рецепторов инозитолтрифосфата (IP3) [17]. Не все наследуемые формы БА объясняются мутациями в вышеупомянутых генах [1]. К дополнительным факторам риска БА следует отнести (по нарастающей) сахарный диабет, курение, низкий уровень образования, гипертонию, депрессию, физическую инертность [15].
Роль астроцитов в болезни Альцгеймера
Глиальные клетки в ГМ поддерживают гомеостаз, выполняют трофическую, опорную, барьерную и секреторную функции. Нарушения в работе глиальных клеток могут в значительной степени способствовать прогрессированию нейродегенеративных заболеваний или даже стать их причиной. При БА в ответ на образование амилоидных бляшек астроциты и микроглия продуцируют широкий спектр провоспалительных факторов, таких как интерлейкин-1 (IL-1) и фактор некроза опухолей α (TNFα), влияя на иммунную систему мозга [18]. Переключение астроцитов на новый функциональный профиль, так называемая активация, нарушает выполнение их основных функций и приводит к гибели нейронов [19, 20, 21]. Активированные астроциты частично фагоцитируют бляшки Aβ, но чрезвычайно медленно и неэффективно [22]. Протофибриллы Aβ42 длительное время хранятся в астроцитах, что вызывает внутриклеточные дефекты, еще сильнее снижающие способность астроцитов к деградации амилоида [22]. По мере прогрессирования БА наблюдаются увеличение количества и размера астроцитов, окружающих амилоидные бляшки, и атрофия астроцитов, находящихся на периферии пораженной зоны, – астроглиоз [23]. Этот процесс включает молекулярные и морфологические изменения, приводящие к увеличению сомы астроцита из-за перепроизводства белков цитоскелета, однако его прерывание снижает нейрональную пластичность и регенерацию ЦНС [21, 24, 25]. При БА также ухудшается способность астроцитов к поддержанию гематоэнцефалического барьера (ГЭБ) и к поглощению глутамата из синаптической щели (рис. 1) в трехстороннем синапсе [26, 27]. Кроме того, исследования, проведенные на трансгенных мышах с БА, показали, что астроциты демонстрируют увеличение концентрации внутриклеточного цитоплазматического кальция ([Ca2+]i) и усиление передачи Ca2+-сигналов [28]. Гиперактивные спонтанные волны астроцитарного кальция, независимые от нейрональной активности и наблюдаемые во многих трансгенных моделях мышей, способны влиять на передачу сигналов в синапсах [23, 28]. Синаптическая дисфункция при БА коррелирует с тяжестью когнитивного спада и ростом числа активированных астроцитов [29]. Таким образом, хотя астроцитарная активация играет защитную роль в головном мозге при БА, реактивные астроциты с нарушенной в них передачей Ca2+-сигналов могут усугублять повреждения нейронов и ускорять прогрессирование заболевания.
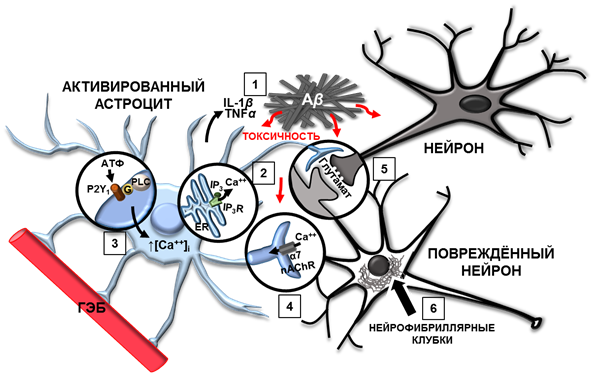
Рис. 1. Изменения в работе астроцитов при болезни Альцгеймера
1. Активированные астроциты продуцируют провоспалительные цитокины: IL-1 и TNFα в ответ на образование Aβ. 2. Передача кальциевых сигналов в астроцитах происходит с помощью вторичного посредника IР3, который связывается с кальциевым каналом на мембранах ЭР и способствует высвобождению ионов Сa2+. 3. Активированные астроциты вблизи амилоидных бляшек экспрессируют метаботропные пуринергические рецепторы P2Y1 до высокого уровня, что приводит к чрезмерному повышению внутриклеточного Сa2+. 4. Aβ индуцирует спонтанное повышение [Сa2+]i в астроцитах гиппокампа посредством α7-никотиновых рецепторов. 5. Пониженная способность к поглощению глутамата у активированных астроцитов влияет на поглощение перисинаптического глутамата, приводя к глутаматной эксайтотоксичности. 6. Внутриклеточные нейрофибриллярные клубки нарушают аксональный транспорт и синаптический метаболизм, приводя к деградации нейрона.
Динамика кальция в астроцитах при болезни Альцгеймера
Ионы кальция – универсальные внутриклеточные посредники, способные регулировать огромное разнообразие внутриклеточных процессов: экзоцитоз, встраивание рецепторов в мембрану, запуск синтеза белков; имеют опосредованное влияние на процессы обучения и памяти [23]. Поступление кальция из внеклеточного пространства происходит в основном через вольтаж-зависимые кальциевые каналы (voltage-gated calcium channel, VGCC), депо-зависимый вход кальция (store-operated Ca2+ entry, SOCE) и Na+/Ca2+ обменники (sodium-calcium exchanger, NCX) [30]. Кальций-связывающие внутриклеточные ферменты имеют различное сродство и чувствительность к ионам Сa2+ (от сотен нМ в цитозоле до концентраций в диапазоне 100–1000 мкМ в ЭР) [23]. Внутриклеточные Сa2+-датчики локализованы в разных частях клетки, поэтому локальные кальциевые градиенты могут независимо друг от друга регулировать отдельные комплексы Сa2+-зависимых процессов [28]. Механизм передачи кальциевых сигналов включает активацию рецепторов, связанных с G-белками, что приводит к вовлечению фосфолипазы C в гидролиз фосфатидил-инозитол-4,5-бисфосфата – фосфолипида клеточных мембран, с образованием вторичных мессенджеров – водорастворимого IР3 и мембранно-связанного диацилглицерола (DAG). IР3 переходит в цитозоль, где связывается со своим рецептором (кальциевым каналом) на мембранах ЭР и способствует высвобождению ионов Сa2+ [31, 32]. Индуцированное G-белками увеличение цитозольного кальция в астроцитах может происходить как в виде колебаний, так и длительным повышением концентрации [30].
Астроцитарный кальций играет ключевую роль не только внутри клетки, но и в межклеточной передаче сигналов в системе «нейроны – глия». Кальциевые сигналы инициируют высвобождение из астроцитов глиотрансмиттеров, таких как пурины (АТФ и аденозин), γ-аминомасляная кислота (ГАМК), D-серин и глутамат. Потенцированный кальцием выброс глутамата из астроцитов способствует модуляции нейротрансмиссии в трехстороннем синапсе [33]. Высвобождение свободного внутриклеточного цитоплазматического кальция в астроцитах может происходить спонтанно или в ответ на выброс нейротрансмиттера [30, 34]. Например, активация пирамидальных нейронов в гиппокампе вызывает повышение уровня [Сa2+]i в астроцитах путем стимуляции метаботропных глутаматных рецепторов (mGluRs), расположенных на астроцитарных отростках [35]. Это активированное состояние может сообщаться соседним астроцитам через щелевые контакты, с помощью высвобождения астроцитарного АТФ или ряда внеклеточных сигнальных молекул [30, 36]. Активность нейронов также может влиять на распространение и продолжительность Ca2+-событий в астроцитах. Это может происходить без какого-либо увеличения частоты (т.е. без появления новых Ca2+-событий), которое, как полагают, является основой астроцитарного ответа на активность нейронов [37]. АТФ, высвобождаемый из астроцитов во время активности нейронов, способен влиять на синаптическую передачу, действуя на пуринергические рецепторы P2X или P2Y [38]. В нормальном мозге астроциты могут высвобождать АТФ в целях регуляции возбудимости интернейронов, воздействуя на рецепторы P2Y1, и тем самым потенцировать ГАМК-ергическую синаптическую передачу [39]. При БА активированные астроциты, вблизи амилоидных бляшек экспрессируют метаботропные пуринергические рецепторы P2Y1 до высокого уровня, что приводит к усилению P2Y1-опосредованной Сa2+-трансмиссии [40] и, следовательно, к кальциевому дисбалансу.
Многие исследования подтверждают значительную роль дисрегуляции кальция в развитии БА. Воздействие Aβ индуцирует спорадические Сa2+-всплески в астроцитах [41]. Аберрантные флуктуации астроцитарного кальция начинаются вблизи бляшек Aβ и могут распространяться на большие расстояния по всей коре головного мозга. В человеческих астроцитах, выделенных из мозга пациентов с БА посмертно, был отмечен абнормальный уровень экспрессии генов, связанных с кальциевыми сигналами и гомеостазом [30]. Также было показано, что Aβ42 и его токсический фрагмент Aβ25-35 вызывают спонтанные Сa2+-всплески в астроцитах, растущих в смешанных астроглиально-нейронных культурах: Aβ-индуцированные колебания [Сa2+]i продолжались в течение длительного времени и приводили к гибели нейронов, которая наступала в течение 24 часов после введения Aβ в культуру, однако подавление [Сa2+]i колебаний предотвращало гибель нейронов [42]. Кроме того, применение пикомолярных концентраций Aβ42 индуцировало спонтанное повышение [Сa2+]i в астроцитах гиппокампа посредством α7-никотиновых рецепторов (α7nAChRs) [42]. Эти рецепторы представляют собой лиганд-зависимые ионотропные каналы, высокопроницаемые для Са2+ [43]. Показанное агонистическое воздействие Aβ на функциональные астроцитарные α7nAChR делает эти рецепторы потенциальной терапевтической мишенью при БА.
Собственная кальциевая активность астроцитов
Астроциты могут генерировать спонтанные Ca2+-всплески, не зависящие от активности нейронов, активации mGluRs или пуринергических рецепторов. Физиологическое значение таких всплесков не было досконально изучено, но эти внутренние сигналы, вероятно, могут влиять на функционирование нейронов, о чем свидетельствуют некоторые исследования [44, 45]. По распространению астроцитарные Ca2+-всплески, или так называемые события, классифицируются на «фокальные» и «обширные» (т.е. занимающие большую часть клетки) [45, 46]. «Фокальные» события — небольшие повышения [Ca2+]i, обычно ограниченные одной подобластью. «Обширными» называют события, при которых более высокие концентрации [Ca2+]i появляются почти одновременно в нескольких смежных субрегионах. «Фокальные» Ca2+-события составляют подавляющее большинство и зависят от выделения медиатора в синаптическую щель соседствующих с астроцитом синапсов. «Обширные» события встречаются реже и в основном являются результатом одиночного потенциала действия в аксоне, проходящем в области, контролируемой данным астроцитом. Однако до сих пор не изучался вопрос о связи между этими двумя различными типами Ca2+-событий и их интеграции в астроцитах [46]. Параметры Ca2+-событий в астроцитах, такие как размах и длительность, можно рассматривать как количество синапсов и временное окно, в котором происходит модуляции синаптической передачи под действием этих событий [37]. При БА эти параметры могут варьировать в зависимости от локализации амилоидных отложений и степени тяжести патологии. Гипотетически Ca2+-события могут управлять переключением астроцитов с физиологического на иммунный профиль и обратно для соблюдения баланса и предотвращения нейродегенерации. Однако на данный момент «язык» астроцитарных Ca2+-событий в литературе практически не изучен [47]. Дальнейшие исследования механизмов пространственно-временной интеграции активности Ca2+ в астроцитах обеспечат более глубокое понимание кальцийзависимой коммуникации между нейронами и астроцитами и позволят пролить свет на пути возникновения патологии БА.
Заключение
Сейчас уже известно, что астроциты не просто поддерживают, но и активно участвуют в обработке нейронной информации в мозге. Это новое восприятие функционирования ЦНС смещает фокус изучения патологии БА только с нейрональных клеток к взаимодействию астроцитов и нейронов. В литературе появляется все больше доказательств того, что астроцитарный вклад в ранние когнитивные нарушения является важным компонентом БА, но на сегодняшний день остается достаточно малоизученным. Прежде всего астроциты реагируют на колебания внутриклеточного кальция, который контролирует сигнальные свойства астроцитов и их способность к обеспечению нейронов. Aβ может нарушать передачу астроцитарных сигналов кальция, что влияет на высвобождение глиотрансмиттеров, которые жизненно важны для связи нейронов и астроцитов, увеличивая вклад последних в патогенез заболевания. Динамика Са2+ представляет собой основу механизма астроцитарной сигнальной передачи и затрагивает почти все характерные особенности, в том числе генетическую причину БА, а ее нарушения приводят к дисфункции астроцитов и подконтрольных им областей головного мозга посредством аберрантных кальциевых сигналов. Расшифровка кальциевых сигналов и поиск соединений, нормализующих дисрегулированные уровни Ca2+ или специфических блокаторов Ca2+-регулируемых патогенных сигнальных каскадов, теоретически могут вернуть физиологическую роль астроцитам и, следовательно, привести к клиническому улучшению.
Библиографическая ссылка
Кушнирева Л.А., Коркотян Э.А. НАРУШЕНИЕ ПЕРЕДАЧИ КАЛЬЦИЕВЫХ СИГНАЛОВ В АСТРОЦИТАХ ПРИ БОЛЕЗНИ АЛЬЦГЕЙМЕРА (ОБЗОР) // Современные проблемы науки и образования. – 2018. – № 4. ;URL: https://science-education.ru/ru/article/view?id=27945 (дата обращения: 19.04.2024).