



Дефект межпредсердной перегородки (ДМПП) – врожденный порок сердца (ВПС), характеризующийся наличием сообщения между правым и левым предсердием, что приводит к артериовенозному сбросу между ними [1; 2]. ДМПП среди всех заболеваний сердечно-сосудистой системы является одним из наиболее распространенных ВПС [3; 4]. По данным различных авторов, частота его встречаемости, по клиническим данным, как изолированная аномалия составляет 5-20%, как часть других аномалий – 30-50%, а по патологоанатомическим – 3,7-10% от всех ВПС [3; 5]. Среди детей данный порок занимает второе место, составляя 8-11% [4; 6]. Появление данного ВПС связано с гипоплазией первичной и вторичной межпредсердной перегородки и эндокардиальных валиков в период эмбриогенеза [1; 7]. Давать начало дисорганогенезу могут генетические, физические, экологические и инфекционные факторы [7]. Дополнительным риском развития ДМПП у ребенка является наличие родственников с ВПС [7]. Кроме наследственной предрасположенности, к формированию ДМПП могут приводить вирусные заболевания (краснуха), эндокринопатии, прием некоторых лекарственных препаратов во время беременности, производственные вредности, осложнения беременности (токсикозы, угроза выкидыша) [7]. Постоянно растет число известных генов, мутации в которыхмогут быть причиной нарушений нормальногоразвития сердца [8]. Доля явно семейныхслучаев составляет около 3% [8]. Изолированные семейные ДМПП обусловлены доминантно наследуемыми мутациями в трех генах: GATA4, MYH6 и неидентифицированном гене, локализованном на коротком плече хромосомы 5 (5р) [8]. Ген, вовлеченный в патогенез септальных дефектов сердца, - MYH6, который кодирует структурный белок, тяжелую альфа-цепь миозина [8]. Описана семья с дефектами ostium secundum с миссенс-мутацией в гене MYH6, приведшей к замене изолейцина на аспарагин [8]. Экспрессию гена MYH6 регулируют два фактора транскрипции: TBX5 и GA1A4; мутации в любом из этих генов также вызывают у человека ДМПП [8]. Хромосомныесиндромы, преимущественно микроделеционные, обнаруживаются примерно у 3% новорожденных с ВПС [9]. У детей с изолированными ВПС доля выявляемых генетическихнарушений существенно меньше [10]. На долю моногенных форм также приходится около3-5% случаев изолированных форм ВПС, количество генов, ответственных за изолированные ВПС, тоже ежегодно пополняется [10]. Наиболее частыми ранними симптомами заболевания при больших ДМПП у детей являются одышка и сердцебиение, но обычно в течение первых месяцев жизни происходят компенсация гемодинамики и регресс клинической картины [1; 7]. Явления недостаточности кровообращения малозаметны и сводятся к умеренно выраженной вялости, потливости, бледности кожных покровов, цианозу носогубного треугольника [1; 3; 4]. Аускультативно порок можно заподозрить при наличии систолического шума во 2-3 межреберьях слева от грудины и расщеплении 2 тона в третьей точке [1; 3; 4]. Ведущим методом диагностики порока является ЭхоКГ с режимом цветного доплеровского картирования [1; 3; 4]. На ЭКГ прослеживается отклонение электрической оси сердца вправо, расширение правых отделов сердца, неполная блокада правой ножки пучка Гиса [1; 3; 4]. Помимо этих исследований, рекомендуется проведение рентгенографии органов грудной клетки для исключения патологии дыхательной системы [1]. Порок достаточно долго протекает относительно благоприятно, но объемная перегрузка правых отделов сердца из-за сброса крови слева направо может со временем прогрессировать, служа причиной нарушения гемодинамики даже у асимптомных больных [4; 7]. Помимо этого, происходит истончение стенок дефекта, что приводит к его постепенному увеличению [4; 6; 7]. Лекарственная терапия при изолированных формах ДМПП не рекомендуется, за исключением наличия у пациентов симптомов сердечной недостаточности и сопутствующих аритмий [1]. Хирургическая коррекция обычно требуется при больших размерах дефекта и при соотношении Qp/Qs более 1,5:1 [5-7]. Проводится чрескожное катетерное закрытие дефекта с помощью кардиологических окклюдеров [3; 5; 6]. Также прибегают к открытым операциям в условиях искусственного кровообращения [1]. Хирургическое лечение изолированных ДМПП не рекомендуется выполнять детям в возрасте до 12 месяцев [1].
Материалы и методы исследования. Из анамнеза известно, что на 20-й неделе беременности при ультразвуковом исследовании выявлен врожденный порок сердца плода с обогащением малого круга кровообращения белого типа – вторичный дефект межпредсердной перегородки. На 32-й неделе - визуализируется истончение межпредсердной перегородки (МПП) на всем протяжении в виде аневризмы, выбухание в полость левого предсердия. Межпредсердное сообщение (МПС) диаметром 4,7 мм, сброс справа налево. Межжелудочковая перегородка (МЖП) интактна. Атривентрикулярные клапаны расположены на одном уровне. Размеры полостей сердца плода: правые отделы дилатированы. Конечный диастолический размер (КДР) левого желудочка (ЛЖ) – 12,8 мм. КДР правого желудочка (ПЖ) – 16 мм. Магистральные сосуды: створки аортального клапана (АоК) утолщены. Дуга аорты на уровне перешейка – 3,5 мм. Наследственность отягощена: известно, что у мамы ВПС - вторичный ДМПП, по поводу которого оперирована в возрасте 4 лет. У бабушки (50 лет) по линии мамы такой же ВПС – вторичный ДМПП с развитием сердечной недостаточности III ст. Ребенок родился от первой беременности, протекавшей на фоне гестоза и анемии легкой степени, вторых срочных самостоятельных родов на сроке 39 недель в головном предлежании. Масса тела при рождении 3190 г, рост 52 см, оценка по шкале APGAR 8/9 баллов. После рождения при осмотре общее состояние ребенка средней степени тяжести, сознание ясное. Кожные покровы и видимые слизистые бледные, чистые. Подкожно-жировой слой развит умеренно. Отеков нет. При аускультации выслушивается пуэрильное дыхание, хрипов нет. ЧД до 42 в мин. Перкуторно границы сердца в пределах возрастной нормы. Аускультативно тоны сердца ясные, ритм правильный. Выслушивается систолический шум во втором и третьем межреберьях слева от грудины, акцент второго тона над легочной артерией. Живот мягкий, не вздут. Печень выступает из-под края реберной дуги на 1 см. Пульсация артерий тыла стопы удовлетворительная с обеих сторон. В 1-е сутки было проведено инструментальное исследование. На ЭКГ (рис. 1) зафиксированы синусовый ритм с ЧСС 120-130 уд./мин., электрическая ось сердца (ЭОС) отклонена резко вправо. Электрическая активность представлена одиночными наджелудочковыми экстрасистолами средней частоты (всего 9).
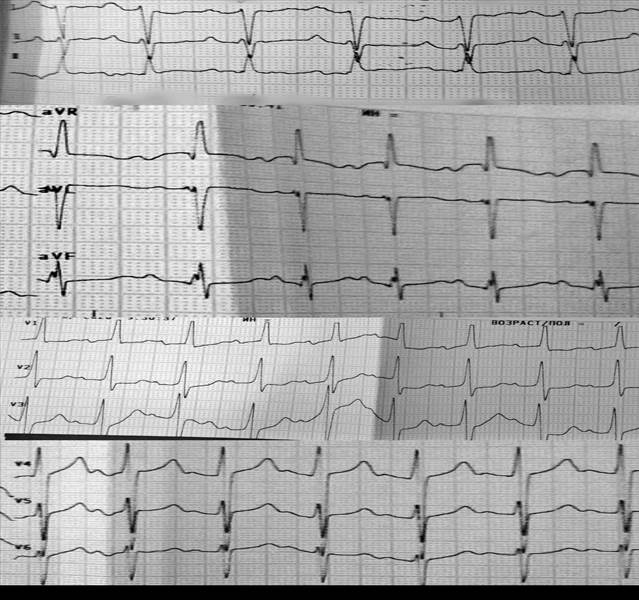
Рис. 1. ЭКГ: синусовый ритм с ЧСС 120-130 уд./мин., электрическая ось сердца отклонена резко вправо. Преобладание электрической активности правого желудочка
При проведении ЭхоКГ (трансторакально) в левых отделах сердца: аорта (Ао) фиброзное кольцо 8,2 мм, восходящий отдел 10,8 мм, проксимальный отдел дуги Ао 6,8 мм, дистальный отдел дуги Ао 5,1 мм, перешеек Ао 4,4 мм, на аортальном клапане створки не изменены. Левое предсердие (ЛП) 12,4 мм (норма 10-15 мм). ЛЖ: КДР 14,9 мм (норма 15-19 мм), конечный систолический размер (КСР) 9,6 мм, конечный диастолический объем (КДО) 6,0 мл, конечный систолический объем (КСО) 1,8 мл, ударный объем (УО) 4,2 мл, фракция выброса (ФВ) 70% (норма 65-75%), сократимость миокарда ЛЖ удовлетворительная, на митральном клапане створки не изменены, регургитация 1 степени. Правые отделы сердца: ПЖ 10,7 мм (норма 3-12 мм), передняя стенка 3,3 мм. Правое предсердие (ПП) 18,4 мм (норма 10-15 мм), на трикуспидальном клапане створки не изменены, регургитация 1 степени. Заключение: центральный дефект межпредсердной пергородки (7,5 мм), дилатация полости правого предсердия, открытый артериальный проток (1,9 мм), сброс слева направо.
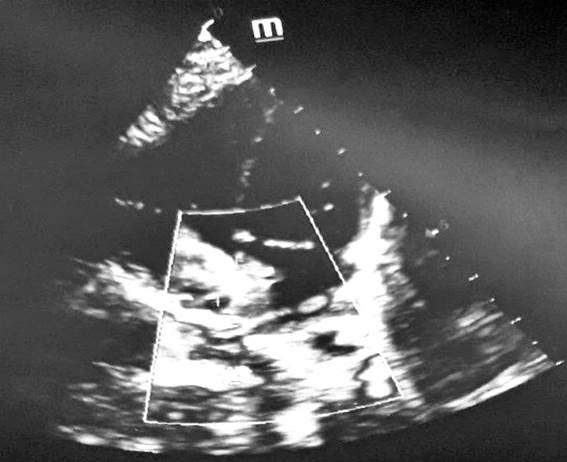
Рис. 2. ЭхоКГ: центральный дефект межпредсердной перегородки, дилатация полости правого предсердия
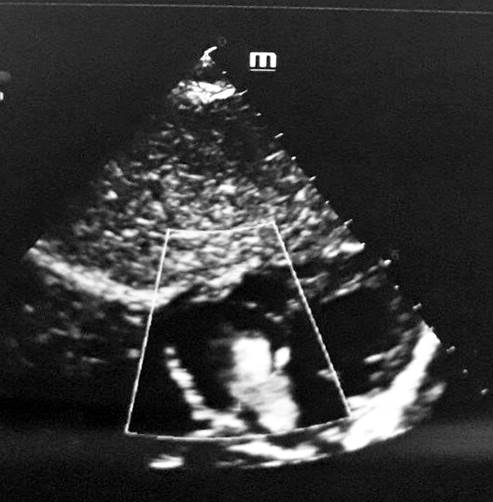
Рис. 3. ЭхоКГ (эпигастральный доступ): вторичный ДМПП, дилатация полости правого предсердия
На ЭХОКГ с доплерографией: фиброзное кольцо легочной артерии 1,8 мм рт. ст., легочная артерия в области ствола 2,2 мм рт. ст., время ускорения потока – АТ 93 м/с, время изгнания – ЕТ 227 м/с, АТ/ЕТ 0,41, среднее давление ЛА 17 мм рт. ст., фиброзное кольцо аорты 3 мм рт. ст., аорта на уровне перешейка 4,5 мм рт. ст., аорта брюшной отдел V max 0,72 м/с. Характер кровотока – магистральный. Расчетное систолическое давление в ПЖ не более 20-21 мм рт. ст. Отношение легочного и системного кровотока - Qp : Qs не превышает 1,5. Заключение: вторичный ДМПП, дилатация полости правого предсердия, открытый артериальный проток. На рентгенографии органов грудной клетки отмечается тимомегалия и расширение сердечной тени. Кардиоторакальный индекс 63%.
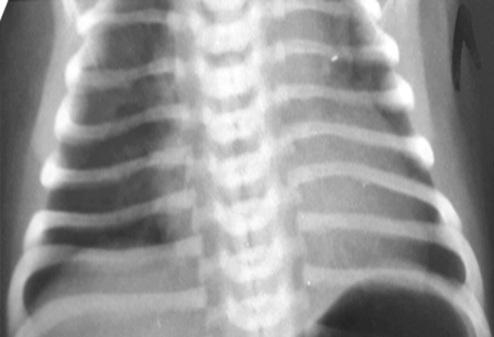
Рис. 4. Рентгенография органов грудной клетки: тимомегалия, сердечная тень расширена, кардиоторакальный индекс 63%
Ребенок был осмотрен кардиологом, впоследствии выставлен диагноз: «врожденный порок сердца с обогащением малого круга кровообращения: вторичный ДМПП с нарушением внутрисердечной гемодинамики. Открытый артериальный проток. Сердечная недостаточность 1-й степени». Учитывая нарушение внутрисердечной гемодинамики, подключили верошпирон в суточной дозе 2 мг/кг.
Результаты исследования и их обсуждение. В связи с кардиомегалией на рентгенологическом снимке, для уточнения размеров ДМПП, ребенок консультирован в Федеральном центре сердечно-сосудистой хирургии г. Пенза, где диагноз был подтвержден. Оперативное лечение в настоящее время не показано, согласно клиническим рекомендациям по ведению детей с ДМПП. Хирургическое лечение изолированных ДМППне рекомендуетсявыполнять детям в возрасте до 12 месяцев [1]. Выписан на 7-е сутки в удовлетворительном состоянии. Рекомендовано наблюдение у педиатра и детского кардиолога по месту жительства. Спонтанное закрытие вторичного ДМПП диаметром 4-5 мм наблюдается у 56% пациентов, 6-7 мм - у 30%, 8-10 мм - у 12% [1]. Размер вторичного ДМПП может меняться с возрастом. У 70% больных вторичный ДМПП ≤ 4 мм уменьшается в размерах, у 12% не меняется и у 18% увеличивается [1]. У нашего пациента есть вероятность спонтанного закрытия ДМПП, но небольшая. С учетом оперативного лечения ДМПП у мамы, естественного течения с неблагоприятным прогнозом у бабушки наблюдение ребенка с проведением динамических ЭхоКГ должно быть индивидуальным. Риск повторения ВПС у ребенка при изолированном ДМПП у матери составляет 4-6%, у отца - 1,5-3,5% [10]. Профилактика возникновения ВПС очень сложна и во многих случаях сводится к медико-генетическому консультированию и разъяснительной работе среди людей, относящихся к группе повышенного риска заболевания [10]. В нашем примере, когда 3 человека, состоящие в прямом родстве, имеют ВПС, вероятность появления следующего случая составляет 65-100%.
Заключение. В среднем изолированные ВПС имеют небольшой риск повторения в семье и оцениваются в 3-4%, но для разных ВПС и семейной отягощенности эти значения буду отличаться, иногда существенно [10]. Хирургическое лечение ВПС должно проводиться в наиболее оптимальные сроки, а не немедленно по выявлении порока, и не в самые ранние сроки, за исключением критических случаев, угрожающих жизни [1; 11]. Лучше всего дети восстанавливаются при операции, перенесённой в возрасте от 2 до 6 лет [1; 11]. Малые дефекты, которые тем не менее не закрылись в детстве, во взрослом и пожилом возрасте могут проявляться симптомами [1; 11]. Так и случилось у бабушки данного ребенка. Несмотря на большое число исследований и имеющихся данных, трудно в каждом случае понять причину ВПС [10]. Во многом это связано с изучением эмбриогенеза человека на ранних стадиях развития. Имеющиеся генетические технологии могут оказать диагностическую помощь многим семьям с пациентами с ВПС [10].