



Среди структурной хромосомной патологии аномалии хромосомы Х наблюдаются в основном у пациентов женского пола, нарушения хромосомы Х у мужчин часто летальны [1]. Как правило, в случаях повреждения одной из хромосом Х, происходит ее инактивация, за счет чего основная функциональная роль переходит к неповрежденной хромосоме Х. При дупликациях хромосомы Х возможны два клинических варианта: отсутствие фенотипических проявлений (обычно при небольших дупликациях) или наличие таковых (при дупликациях большего размера) [2, 3, 8, 12, 16]. Как указывают многие наблюдения, различия фенотипических особенностей в зависимости от размера дупликаций связаны с тем, что обширные дуплицированные участки хромосом, находясь в инактивированной хромосоме Х, сохраняют экспрессию генов, в отличие от небольших по размеру дупликаций. В данной работе мы представляем два случая крупных дупликаций большей части длинного и короткого плеч хромосомы Х у девочек с тяжелыми клиническими проявлениями.
Цель работы
Целью работы явилось выявление и уточнение хромосомной патологии молекулярно-цитогенетическими методами, исследование особенностей инактивации пораженной хромосомы Х в случаях дупликаций участков Xp11.2-> pter и Xq21.3-> q28 на хромосоме Х и связи с клиническими проявлениями.
Mатериалы и методы
Цитогенетические исследования проводились на хромосомах лимфоцитов периферической крови, культивированных стандартным методом с применением GTG и CBG окрашивания [9]. Молекулярно-цитогенетические исследования были проведены методом FISH с использованием ДНК пробы MCB (Multicolor chromosome banding) [6] и сайтспецифичных ДНК проб, включавших пробу на ген MECP2, участки Хpter и Xqter из коллекции лаборатории генетики и геномики психических заболеваний НЦПЗ РАМН [10,14]. Определение особенностей инактивации хромосом Х проводилось на хромосомах, культивированных с 5-бром-2-дезоксиуридином (BrdU), введенным в клеточную культуру за 6 часов до конца культивирования, по репликационному рисунку хромосом, окрашенных красителем Hoechst 33258 с применением центромерной ДНК пробы на хромосому Х [15].
Pезультаты исследований
Мы приводим описания двух случаев.
Случай 1. Девочка в возрасте 5,5 лет имела следующие клинические проявления: грубую задержку физического, психомоторного и психоречевого развития, микроцефалию, атонически-астатический синдром, поражение зрительных проводящих путей обоих глаз, гипертелоризм глазных щелей, гипертрофию десен, короткую шею, клинодактилию, арахнодактилию, поперечную складку на обеих ладонях, изменения на МРТ по типу адренолейкодистрофии, эпилепсию, спленомегалию, очаги депигментации и гиперпигментации на коже.
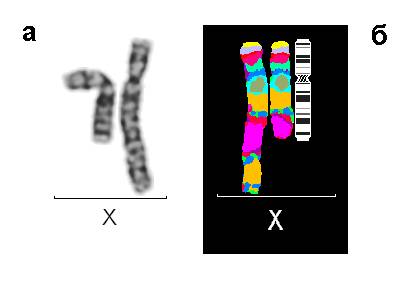
Рис. 1. Результаты исследований пациента 1: (а) Нормальная и аномальная хромосомы Х после цитогенетического анализа, GTG-окраска; (б) Результат флюоресцентной гибридизации с MCB ДНК зондом на хромосому Х
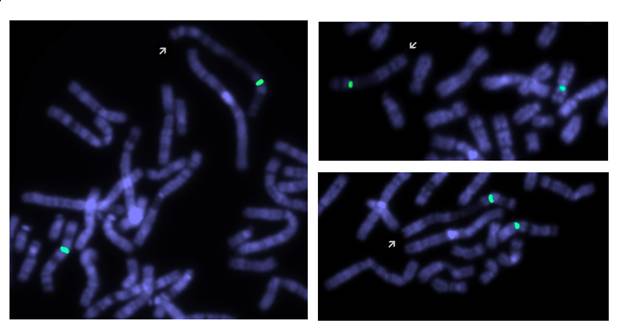
Рис 2. Результаты исследования паттерна репликации пациента: зеленый сигнал маркирует центромеру хромосомы Х. Аномальная хромосома (указана стрелкой) демонстрирует рисунок поздней репликации. Светло окрашенные участки соответствуют наиболее рано реплицирующимся участкам (до введения BrdU).
При цитогенетическом исследовании у девочки был обнаружен дополнительный материал на длинном плече хромосомы Х, определенный по GTG-окраске, как инвертированная дупликация длинного плеча хромосомы Х. Помимо этого был выявлен дополнительный клон клеток с кариотипом 45,Х. Молекулярно-цитогенетическое исследование с MCB пробой на хромосому Х и ДНК пробой на ген MECP2 позволило уточнить аномалию, как инвертированную дупликацию с частичной трипликацией длинного плеча (рис. 1). Кариотип пробанда после проведенных исследований следующий: 46,X,der(X)dup(q21.3q28)trp(q21.3q25)[57]/45,X[43]. Аномальная хромосома Х была инактивирована в 100% клеток (рис. 2). Кариотипы родителей были нормальные.
Случай 2. Девочка в возрасте 10 месяцев имела грубую задержку психомоторного и психоречевого развития, врожденный порок сердца, эпилепсию, эпикант, аномальный разрез глазных щелей, запавшую переносицу, диспластичные ушные раковины, дополнительный сосок. Цитогенетическое исследование выявило дополнительный материал неизвестного происхождения на длинном плече хромосомы Х. После проведения молекулярно-цитогенетического исследования с MCB пробой на хромосому Х и сайтс-пецифичными ДНК пробами, было определено, что данный материал является коротким плечом хромосомы Х (рис. 3). Мозаицизм в этом случае отсутствовал. Кариотип пробанда был следующим: 46,X,der(X)dup(X)(p11.2pter),9ph или полная запись кариотипа: 46,X,der(X)(Xpter->Xq28::Xp11.2->Xpter),9ph.
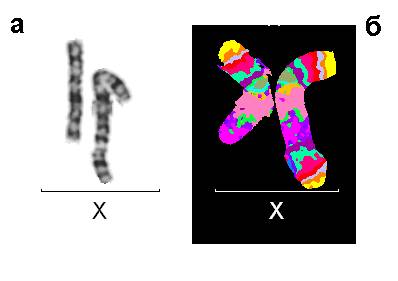
Рис. 3. Результаты исследований пациента 2: (а) нормальная и аномальная хромосомы Х после цитогенетического анализа, GTG-окраска; (б) Результат флюоресцентной гибридизации с MCB ДНК зондом на хромосому Х.
При исследовании особенностей инактивации хромосом Х, было выявлено 8% клеток, где аномальная хромосома была активной (рис. 4). Кариотипы родителей были нормальными: 46,ХХ и 46,ХY,9ph. В обоих представленных случаях ген MECP2, расположенный в участке Хq28, присутствовал в аномальных хромосомах.
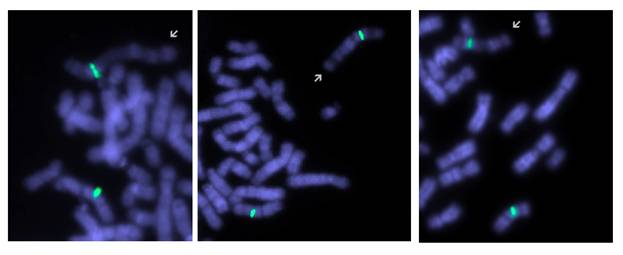
Рис. 4. Результаты исследования паттерна репликации пациента 2: зеленый сигнал маркирует центромеру хромосомы Х. Аномальная хромосома (указана стрелкой) демонстрирует рисунок поздней репликации. Светло окрашенные участки соответствуют наиболее рано реплицирующимся участкам (до введения BrdU).
Обсуждение
В настоящее время в литературе описано несколько десятков случаев дупликации хромосомы Х у девочек [7, 8, 11, 16]. Такие случаи рассматриваются по размерам и генонасыщенности дуплицированного участка, степени инактивации аномальной хромосомы в клетках, клиническим проявлениям у пациентов и наличию или отсутствию мозаичной формы хромосомной патологии. В наших двух случаях дупликации были больших размеров: в первом случае дуплицировано было почти все длинное плечо с частичной трипликацией проксимального участка, а в другом случае дупликация, которая локализовалась на конце длинного плеча, была размером, практически составлявшим все короткое плечо хромосомы Х. В обоих случаях у пациентов наблюдались тяжелые клинические проявления, несмотря на то, что аномальная хромосома Х была инактивирована в 100% клеток в случае 1, и в 92% в случае 2. В первом случае обнаруженный мозаицизм 46,X,der(X)[57]/45,X[43], вероятно, мог частично облегчать симптомокомплекс больной. Возможно, клон 45,Х, характерный для синдрома Шерешевского-Тернера, в данном случае, как ни парадоксально, оказывал положительное влияние на фенотип. Задержка физического развития у этой пациентки могла быть одним из проявлений синдрома Шерешевского-Тернера, хотя общая клиническая картина была значительно тяжелее и данному синдрому не соответствовала.
Суммируя полученные нами данные, можно сделать с большой вероятностью вывод о том, что дополнительные фрагменты хромосомы Х не были инактивированы, вызывая тяжелые клинические проявления. Последние клинические наблюдения и лабораторные исследования показали, что Х-инактивации подвержены в основном дуплицированные фрагменты малого размера. Известно, что не все гены в хромосоме Х инактивируются. Около 10-15% генов не подвергаются инактивации[4]. Эти гены распределены по длине хромосомы Х, а также в псевдоаутосомных участках (PAR), расположенных в терминальных регионах короткого и длинного плеч, и в участках, гомологичных хромосоме Y. Существует мнение о том, что негативные клинические проявления в случаях Х-дупликаций вызваны влиянием дополнительной дозы этих генов [5]. Однако тяжесть клинических проявлений у пациентов с подобными перестройками хромосомы Х свидетельствует не в пользу этого мнения. В наших случаях анализ репликационного рисунка на хромосомах Х показал, что участки дуплицированного материала, наиболее удаленные от центра Х-инактивации, демонстрируют рисунок, характерный для хромосом с ранней репликацией (рис. 2 и 4), что позволяет сделать предположение о транскрипционной активности генов в этих участках. Изменения рисунка репликации в дуплицированных участках наблюдались и другими исследователями [13]. Эти наблюдения являются подтверждением результатов, характерных для аутосомных фрагментов, транслоцированных на хромосому Х [12]. Вероятно, существует общий механизм, благодаря которому, дополнительный материал на инактивированной хромосоме Х остается активным. Как известно, процесс инактивации хромосомы Х осуществляется за счет некодирующей XIST РНК, продуцирующейся центром инактивации XIST в участке Xq13.2 и покрывающей всю хромосому Х. Возможно, случаи дополнительного материала на хромосоме Х имеют связь с дозой XIST РНК. Нельзя также исключать тканеспецифических особенностей характера инактивации аномальной хромосомы Х.
Заключение
Таким образом, полученные нами данные указывают на то, что обширные дуплицированные участки, состоящие из материала короткого или длинного плеч хромосомы Х, расположенные в дистальной ее части, сохраняют свою транскрипционную активность, тогда как сама перестроенная хромосома подвергается инактивации. Для более полного понимания эпигенетических механизмов, за счет которых дополнительные фрагменты ДНК хромосомы Х могут не инактивироваться, требуются дальнейшие молекулярные и молекулярно-цитогенетические исследования.
Исследование выполнено за счет гранта Российского Научного Фонда (проект №14-15-00411).