



Среди обширного спектра хромосомных и геномных аномалий, обнаруживаемых у детей с задержкой психоречевого и полового развития, видное место занимают различные численные и структурные аномалии половых хромосом (гоносом). Наиболее изучены и часто встречаются синдромы Шерешевского-Тернера (кариотипы - 45,Х; 46,X,i(Xq); 45,X/46,XХ и другие), Клайнфельтера (кариотипы - 47,XXY; 48,XXXY и другие), трисомии хромосомы Х (кариотип - 47,ХХХ) и дисомии хромосомы Y (кариотип - 47,XYY) [2; 15]. Наблюдаются как регулярные, так и мозаичные формы этих синдромов; возможен тканевой мозаицизм. Клинические признаки пациентов сильно варьируют от почти полного их отсутствия, особенно в случаях мозаицизма с малой долей аномального клона, до выраженной умственной отсталости, пороков и микроаномалий развития, нарушения репродуктивных функций и других симптомов. Диагностика подобных случаев нередко, особенно при мозаицизме и структурных перестройках, требует применения таких молекулярно-цитогенетических методов исследований, как флюоресцентная гибридизация in situ (FISH) с хромосомоспецифичными и сайтспецифичными ДНК зондами [10], метафазная сравнительная геномная гибридизация, а также серийная сравнительная геномная гибридизация на ДНК-микроматрицах (array CGH), что позволяет уточнить генетический диагноз и проводить корректное медико-генетическое консультирование семей [3; 4; 7; 11].
Частота встречаемости синдрома дисомии хромосомы Y – 1-1,5:1000 новорождённых [1; 2; 14], а среди мужчин с психическими отклонениями пациенты с дисомией хромосомы Y встречаются в 0,45-15% случаев [15]. В основном дисомия хромосомы Y возникает в результате неправильного расхождения хромосом в отцовском мейозе II [16]. По данным литературы, при синдроме дисомии хромосомы Y отсутствуют обязательные признаки, характерные для гоносомных синдромов, но среди необязательных признаков часто отмечают высокий рост, умственную отсталость различной степени тяжести, нарушение половой дифференцировки (крипторхизм, гипогонадизм, дисплазия гениталий); у примерно 30% мужчин с этим синдромом отмечено нарушение репродуктивной функции [13], агрессивное, иногда асоциальное поведение, психопатические черты характера (импульсивность, отсутствие сильных привязанностей, плохое владение собой по поводу примитивных эмоций), черты аутизма; у некоторых больных выявляют шизофрению, депрессивные психозы, тяжёлые формы психопатии и эпилепсии. Среди других аномалий отмечают макроцефалию, прогнатизм, выступающие надбровные дуги, высокое нёбо, гипертрофию языка, увеличение конечностей. Показано также, что большинство опубликованных случаев данного синдрома обнаруживают во взрослых и детских психиатрических лечебницах и лечебно-профилактических учреждениях для содержания социально опасных пациентов, а также в тюрьмах [2; 5]. В целом для синдрома дисомии хромосомы Y характерно разнообразие клинических проявлений, что представляет большой интерес для клинических генетиков и заставляет тщательно исследовать каждый такой случай с целью накопления данных и установления корреляции «фенотип-генотип». Мы представляем результаты обследования ребенка мужского пола с дисомией хромосомы Y, имеющего клинические проявления, не совсем характерные для этого синдрома.
Материалы и методы
В работе обследовался мальчик в возрасте 3 лет. Культура лимфоцитов периферической крови, приготовление препаратов, дифференциальное окрашивание хромосом по длине и анализ кариотипа проводились по стандартным методикам (GTG- и CBG-окрашивание хромосом по длине) [2]. Кроме классического цитогенетического анализа, также было проведено молекулярно-цитогенетическое исследование – флюоресцентная in situ гибридизация (FISH) с центромерными хромосомоспецифичными ДНК-зондами на хромосомы Х и Y из оригинальной коллекции лаборатории [10; 18; 20]. Гибридизация и анализ препаратов проводились по стандартным протоколам [17; 19].
Результаты
В клинику института для обследования поступил ребёнок 3 лет, родившийся от 2-й беременности, 2-х физиологических родов с массой тела 3750 г, длиной - 54 см. От 1-й беременности родился здоровый мальчик, ему 5 лет. Причиной обращения послужили следующие клинические признаки: задержка психоречевого развития и аномалии поведения. При осмотре ребёнка отмечены такие симптомы, как ограниченность понимания обращённой речи, несформированность навыков опрятности и самообслуживания, стереотипные действия с предметами неигрового назначения, резко отрицательная реакция на запреты и ограничения в виде падения на пол и ударов головой об пол, а также агрессии к окружающим (укусы и т.д.), ходьба на цыпочках. Кроме того, у пациента обнаружены хронические кататоно-аффективные расстройства, аллергические реакции на пыль, плесень и шоколад, дисфункция желчного тракта, плоско-вальгусная деформация стоп, пролапс митрального клапана. Из родословной (рис. 1) видно, что прапрадеды пробанда по отцовской и материнской линиям являются родными братьями, и таким образом, родители пробанда – троюродные сибсы. В родословной, со слов родителей, не выявлено случаев умственной отсталости или пороков развития, у деда по отцовской линии обнаружен поллиноз, у деда по материнской линии – ишемическая болезнь сердца (ИБС).
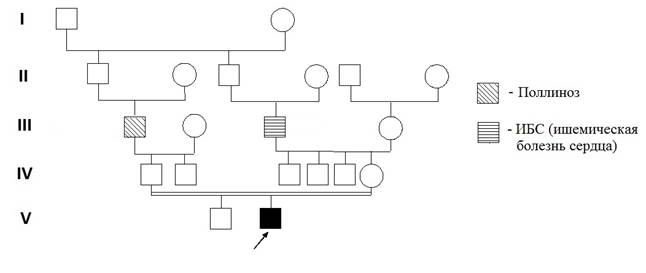
Рис. 1. Родословная пациента с синдромом дисомии хромосомы Y
На основании клинических признаков принято решение о проведении цитогенетического исследования. Из анализа кариотипа (рис. 2) видно, что у пробанда выявлены две хромосомы Y. В результате проведённых цитогенетических исследований кариотип пробанда (GTG- и CBG-окрашивания хромосом по длине) - 47,XYY. Других численных или структурных аномалий хромосом, обнаружимых стандартными цитогенетическими методами, у данного пациента не выявлено.
Для исключения мозаицизма проведено FISH исследование (рис. 3), при котором не обнаружено предполагавшегося хромосомного мозаицизма гоносом, и дисомию хромосомы Y следует считать регулярной и возникшей de novo: 47,XYY. ish (DXZ1x1;DYZ3x2) [100]. nuc ish (DXZ1x1;DYZ3x2) [1000]. Таким образом, после проведения цитогенетического и молекулярно-цитогенетического исследований ребёнку поставлен диагноз – синдром дисомии хромосомы Y.
На основании результатов цитогенетических и молекулярно-цитогенетических исследований с учётом родства родителей принято решение о дополнительном проведении пробанду молекулярно-цитогенетического исследования – серийной сравнительной геномной гибридизации (молекулярного кариотипирования или array CGH) [4; 8; 11]. Проведение молекулярного кариотипирования данному пациенту позволит выявить возможные аномалии генома, а также установить участки потери гетерозиготности (loss of heterozygosity, LOH), которые в данном случае ввиду родства родителей (троюродные сибсы) могут быть обширны и способны значительно (негативно) повлиять на клинические проявления у пробанда [12].
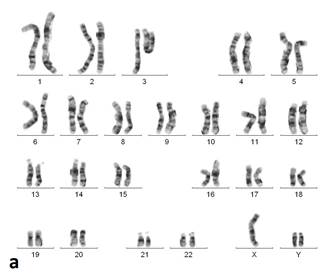

Рис. 2. Кариотип пробанда после проведения GTG- (а) и CBG- (б) окрашивания (47,XYY)
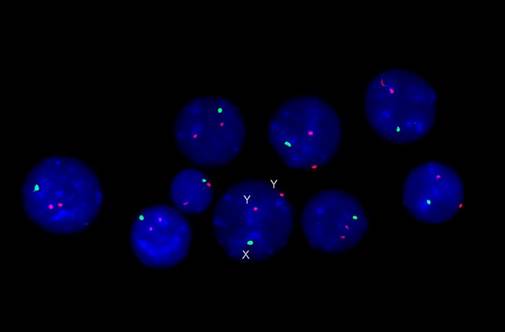
Рис. 3. Результаты FISH-исследования с центромерными ДНК-зондами на хромосомы Х и Y (зеленые сигналы – хромосома Х, красные – хромосома Y)
На рисунке представлены интерфазные ядра лимфоцитов периферической крови, в каждом из которых по две хромоcомы Y.
Обсуждение
В достаточно широком спектре различных численных и структурных аномалий половых хромосом (гоносом) дисомия хромосомы Y занимает особое место благодаря исключительной вариабельности клинических признаков данного синдрома: от почти полного отсутствия каких-либо патологических проявлений до умственной отсталости тяжёлой степени; бесплодие наблюдается лишь в 30% пациентов, и прежде всего это связано с нарушением сперматогенеза. Причины клинического полиморфизма при этом синдроме не всегда ясны и могут быть связаны с иными хромосомными (геномными) микроаномалиями, не обнаружимыми стандартными методиками классического кариотипирования [4; 7].
Данный случай необычен тем, что у пациента с регулярной дисомией хромосомы Y отмечены выраженные черты аутизма, что при данной патологии наблюдается нечасто [4]; вероятно, на картину заболевания повлияло и то, что ребёнок рождён в кровнородственном браке (родители – троюродные сибсы и принадлежат к одному и тому же этносу (аварцы) Северного Кавказа, где кровнородственные браки у некоторых народов нередки); поэтому у пробанда возможно наличие обширных участков потери гетерозиготности (LOH) более чем в 3% генома (коэффициент инбридинга 1/32) [6; 12]. Именно молекулярное кариотипирование и биоинформатический анализ in silico позволит выявить данные участки, что при их наличии может привести и к возникновению различных наследственных аномалий [8; 9]. С учётом всех данных этой семье рекомендовано медико-генетическое консультирование с возможной пренатальной диагностикой при повторном деторождении.
Заключение
В данной статье мы рассматриваем необычный случай синдрома дисомии хромосомы Y у мальчика, рождённого в кровнородственном браке. Изучение влияния на клинические проявления у пробанда как регулярной формы дисомии хромосомы Y, так и кровнородственного брака весьма важно для понимания вариабельности клинических проявлений синдрома дисомии хромосомы Y. Проведение array CGH (молекулярного кариотипирования на ДНК-микроматрицах) с последующим биоинформатическим анализом позволит выявить возможные дополнительные микроаномалии генома, не обнаруженные классическими цитогенетическими методами, а также участки потери гетерозиготности (LOH), которые тоже могут влиять на фенотип [8; 9].
Накопление данных о подобных случаях позволит более эффективно устанавливать корреляцию «фенотип-генотип», корректно описывать особенности фенотипа при различных хромосомных синдромах и проводить медико-генетическое консультирование семей с ребёнком, рождённым в кровнородственном браке.
Исследование выполнено при финансовой поддержке Российского научного фонда (проект № 14-15-00411).