



Несовершенный остеогенез (НО) – это группа наследственных заболеваний скелета, связанная с врожденными генерализованными нарушениями соединительной ткани, характеризующимися тяжелым остеопорозом и хрупкостью костей. В зависимости от типа НО может наблюдаться такая симптоматика и признаки, как задержка умственного развития, несовершенный дентиногенез, голубые склеры, потеря слуха, низкий рост, сколиоз, гипермобильность суставов, гиперэластичность кожи и связок и др. [8].
Распространенность и частота заболевания по данным различных авторов варьирует в широких пределах. Так, по данным Carter C.O. (1977) [5], частота заболевания составляет 0,4 на 10000 новорожденных. Распространенность заболевания, по данным Orfanet (по состоянию на ноябрь 2016 г.) [9], составляет 10,0 на 100000 населения.
В российских популяциях распространенность НО с аутосомно-доминантным (АД) типом наследования составляет: в республиках Адыгея (1:65000), Саха Якутия (1:7869), Башкортостан (1:24824), Татарстан (1:52316), Удмуртия (1:22194), Марий Эл (1:57190), Костромская (1:74080), Тверская (1:25333) области [1,2].
В настоящее время, в соответствии с Международной номенклатурой наследственных заболеваний скелета 2015 г. [4], выделяют пять основных типов несовершенного остеогенеза (НО), каждый из которых объединяет в себе группу моногенных заболеваний с преимущественно схожей клинической картиной: недеформирующая форма НО, тип 1 (АД тип наследования), пренатальная летальная форма НО, тип 2 (АД, аутосомно-рецессивный (АР) тип наследования), постепенно деформирующий тип НО, тип 3 (АД, АР), умеренная форма НО, тип 4 (АД, АР), НО с кальцификацией межосных мембран и/или гипертрофированными костными мозолями, тип 5 (АД).
По данным Online Mendelian Inheritancein Man (OMIM), 2017 г. [7] с учетом генетической гетерогенности различают 17 вариантов (типов) изолированного НО. Как основное проявление, либо как один из ведущих симптомов заболевания встречается в 87 нозологиях.
Цель исследования. Показать важность детального мультидисциплинарного подхода к диагностике наследственных остеохондродисплазий ввиду возможного выраженного внутрисемейного клинического полиморфизма на примере описания клинического случая несовершенного остеогенеза, сочетающегося с новообразованиями цемента зуба у пациентов одной семьи, а также его влияние на изменение показателей распространенности и отягощенности наследственных остеохондродисплазий в Ростовской области, в частности, несовершенного остеогенеза, при адекватной дифференциальной диагностике клинически схожей патологии.
Материалы и методы исследования
Настоящее медико-генетическое обследование населения 12 районов Ростовской Области (РО): Цимлянский, Волгодонской, Егорлыкский, Целинский, Матвеево-Курганский, Родионово-Несветайский, Миллеровский, Тарасовский, Дубовский, Зимовниковский, Красносулинский и Мясниковский проведено в 2006 г. согласно протоколу (возможность выявления 3000–3500 различных наследственных болезней) генетико-эпидемиологических исследований, разработанному в ФГБНУ «Медико-генетический научный центр» (ФГБНУ «МГНЦ») [1,2]. Суммарная численность обследованного населения составила 497,460 тыс. человек (основное население области более чем на 90 % представлено русскими). Все пациенты осмотрены генетиком, педиатром, неврологом, ортопедом и офтальмологом для выявления сопутствующей патологии. В дальнейшем, согласно методологии протокола, из общего числа выявленных больных были выделены пациенты с наследственными остеохондродисплазиями (НОХД). Для диагностики НОХД, помимо данных клинического осмотра, оценивали результаты рентгенологических методов исследования, ультразвукового исследования (УЗИ) крупных суставов, магнитно-резонансной томографии (МРТ) и рентген-компьютерной томографии (РКТ) различных сегментов и отделов опорно-двигательного аппарата. Весь спектр диагностированных НОХД распределяли в соответствии с Международной номенклатурой наследственных заболеваний скелета 2015 [4].
Результаты и обсуждение
В рамках проведенного медико-генетического исследования выявлен широкий спектр моногенной наследственной патологии (МНП), сопровождающейся различными нарушениями со стороны опорно-двигательного аппарата.
Нозологический спектр НОХД составил 110 заболеваний – 40 заболеваний с изолированным поражением опорно-двигательного аппарата и 70 нозоформ, входящих в состав наследственных синдромов (472 больных). Симптомы нарушения опорно-двигательного аппарата отмечались у 31,87 % пациентов с МНП. Из них пациенты с изолированным поражением опорно-двигательного аппарата (279 больных) составили 18,84 % от общего числа больных с МНП, с наследственными синдромами (193 пациента) – 13,03 %. Соотношение изолированных и синдромальных форм в группе больных с НОХД (472 пациента из 317 семей) составило 59,11 % (279 больных) и 40,89 % (193 больных) соответственно. Среди широкого разнообразия выявленных нозологических форм с признаками НО выявлено 19 семей (29 больных) с АД типом наследования и 1 семья (1 больной) с АР типом наследования. Общая распространенность НО заболевания составила 1:16582 (или 6,03 на 100000 населения).
Для большинства пациентов с диагностированным НО были характерны такие симптомы, как некоторое укорочение и искривление конечностей, саблевидная деформация костей голени, кифосколиоз, деформации грудной клетки, тонкая, легкоранимая кожа, умеренная гипермобильность суставов. Одним из патогномоничных признаков были голубые склеры. Умственное развитие у всех больных в пределах возрастной нормы. В анамнезе – частые переломы, возникающие от незначительной травмы, при этом тяжесть переломов не соответствовала степени травмы. При рентгенологическом обследовании определялись косвенные признаки остеопороза, истончение кортикального слоя, тонкие диафизы с расширенными метафизами, консолидированные переломы и костные мозоли, изменения со стороны тел позвонков (двояковогнутые формы). Данная симптоматика позволила предположить у больных с АД типом наследования наличие НО тип I.
В дальнейшем, при тщательном анализе полученных клинических, анамнестических и генеалогических данных был выявлен интересный случай, с первично диагностированным НО, сочетанным с доброкачественной опухолью нижней челюсти (по данным предоставленной на момент осмотра медицинской документации – цементома) и аномалиями длинных трубчатых костей (хрупкость/множественные переломы и изогнутость большеберцовых костей) (1 семья, 3 больных). У членов семьи было взято письменное информированное согласие на участие в данном исследовании. По данным различных авторов [3,6,11,12,13], данное сочетание скелетных аномалий является крайне редким и требует дифференцировки между такими заболеваниями, как гнатодиафизарная дисплазия (GDD, OMIM#: 166260), гигантоформная цементома (GC, OMIM#: 137575), и McCune-Albright синдром (MAS или FD, OMIM#: 174800). Описание клинических случаев FD и GDD, а также их дифференциальная диагностика в литературе освещены крайне скудно [3,10,12]. По данным Е.И. Рогаева и соавт. 2016 г. [3], множественные патологические переломы в детском и подростковом возрасте одинаково характерны для FD и GDD. Дифференциальная диагностика их возможна при рентгенологическом исследовании. Наличие утолщения кортикального слоя в зоне диафиза и его искривление специфично для GDD. Интересно, что при GDD непосредственно процесс консолидации переломов не нарушен. При нем отсутствуют явления превдоартрозов и значимые деформации длинных трубчатых костей в зоне консолидированного перелома. Для внескелетных проявлений GDD характерно отсутствие изменений со стороны кожи и эндокринных нарушений, что позволяет дифференцировать ее от MASсиндрома. В описанных до настоящего времени семейных случаях GC часть членов семей имели переломы и схожую с GDD клиническую картину. Другая их часть – не имели переломов в анамнезе.
В обследованной нами семье оба пробанда имели нетипичную для НО схожую клиническую картину, которая была представлена выраженными саблевидными деформациями костей обеих голеней и незначительной деформацией костей предплечий, вследствие множественных переломов, возникавших при незначительных бытовых нагрузках (приседания, подъем по лестнице и т.п.), наличием в анамнезе у обоих новообразований (цементом) верхней и нижней челюсти. Оба пробанда неоднократно прооперированы в Российском онкологическом Научном Центре им. Н.Н. Блохина (удаление опухоли, протезирование нижней челюсти), где первично (в 1993 г.) был выставлен и гистологически подтвержден диагноз: Гигантоформная цементома. Данный клинический случай был описан В.В. Рогинским и соавт., (2010) [12]. Старший брат оперирован по поводу цементомы (1993 – субтотальная резекция нижней челюсти, 1998 – рецидив опухоли и повторное вмешательство, 2004 – резекция опухоли верхней челюсти). В связи с множественными патологическими переломами, перемещался, используя ортопедическую трость. Младший брат передвигался только в инвалидной коляске, имел в анамнезе большее количество патологических переломов костей, дважды оперирован по поводу цементомы (2007 – субтотальная резекция нижней челюсти, 2009 – резекция опухоли верхней челюсти). Авторы сообщают, что за период наблюдения по поводу новообразований отмечалось прогрессирование деформаций грудопоясничного отдела позвоночника и длинных трубчатых костей, наличие патологических переломов предплечий и голеней у обоих пробандов. Также, у пробандов не было выявлено каких-либо эндокринных нарушений и нарушений костного метаболизма. В связи с недостаточностью материально-технического оснащения, генетические исследования для дифференцировки и уточнения диагноза, в частности на наличие мутации в генах GNAS1 и TMEM16E, не проводились. Кроме того, в статье представлена не полная родословная семьи – только два пораженных сибса, что было связано с психологической замкнутостью членов семьи и значительно затрудняло точную диагностику заболевания.
На момент нашего осмотра у старшего брата (Рис. 1, III-1) наблюдалась следующая клиническая картина: мальчик, нормального роста, с сохраненным интеллектом, без эндокринных нарушений и признаков соединительнотканной дисплазии, «саблевидная» деформация голеней, имеет менее выраженные деформации и более социально адаптирован (передвигается при помощи ортопедической трости), чем его брат. Патологических переломов – 12. Нижняя челюсть протезирована после удаления цементомы.
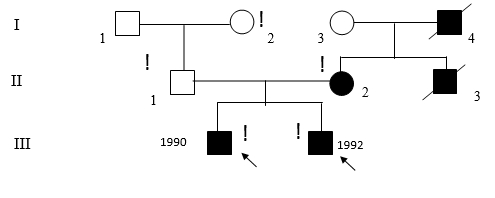
Рис. 1. Родословная семьи с гнатодиафизарной дисплазией
У младшего брата (Рис. 1, III-2) выявлена более тяжелая клиническая картина: мальчик, нормального роста, с сохраненным интеллектом, без эндокринных нарушений и признаков соединительнотканной дисплазии, общее число патологических переломов – 14. Передвигается только с посторонней помощью в кресле-каталке. На рентгенограммах отмечаются множественные переломы костей голени, их грубая «саблевидная» деформация, остеопения костей голени и избыточные костные мозоли, а также переломы костей предплечья (Рис. 2). Нижняя челюсть также протезирована.
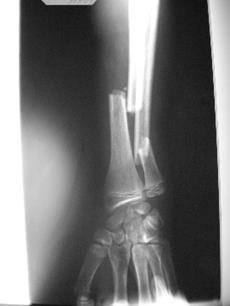
Рис. 2. Переломы костей предплечья у пробанда Рис. 1, III-2
При беседе с бабушкой (Рис. 1, I-3) и отцом (Рис. 1, II-1) пробандов составлена родословная и получена дополнительная информация о её муже, детях, в том числе матери и дяди пробандов. У отца (II-1) пробандов отмечались незначительные признаки соединительно-тканной дисплазии. Дед (I-1) и бабка (I-2) клинически здоровы.
Со слов бабушки, у её мужа были частые переломы, но не более 10, опухолей лицевого скелета не наблюдалось, умственно сохранен. Сын умер в возрасте 26 лет (Рис. 1, II-3). По согласованию с бабушкой пробандов просмотрена его медицинская карта, из которой следовало, что у него опухолей лицевого скелета не наблюдалось, а на основании частых переломов (15-17) поставлен диагноз: НО. Деформаций конечностей специфичных для пробандов («саблевидная» деформация голеней) не было.
У дочери (Рис. 1, II-2) переломы (6–7 в анамнезе) не сопровождались искривлением конечностей (рис. 3).
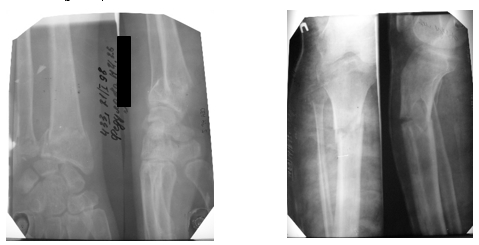
Рис. 3. Рентгенограммы костей голени и коленных суставов матери пробандов
Передвигается самостоятельно, без посторонней помощи. Была осмотрена лишь визуально, от детального клинического осмотра отказалась. Нижняя челюсть протезирована, прооперирована по поводу удаления цементомы, гистологически опухоль идентична таковой у обоих пробандов. Учитывая небольшое количество переломов, отсутствие деформаций конечностей, в 1994 г. ей был выставлен только диагноз: Цементома (черубизм).
Таким образом, всем пораженным членам данной семьи врачами различного профиля были выставлены различные диагнозы (на основании предоставленной медицинской документации): двум пробандам – гигантоформная цементома, их матери – черубизм, дяди - несовершенный остеогенез, больной дед по материнской линии за медицинской помощью по поводу переломов не обращался, лечился самостоятельно. С учетом данных анамнеза, генеалогического обследования, на основании полученных при объективном осмотре пробандов данных нами первично был установлен диагноз: семейная гигантоформная цементома (GC, OMIM#: 137575).
Проведенное Е.И. Рогаевым с соавт. 2016 г. [3] полноэкзомное исследование (ExoME), показало наличие гетерозиготной миссенс мутации c.1067G>A (p.Cys356Tyr) в гене ANO5 у обоих пробандов и первичный диагноз GC, был изменен на гнатодиафизарную дисплазию (GDD). Также, при ExoME-исследовании у обоих пробандов были выявлены еще две редкие гетерозиготные мутации в гене коллагена COL5A1 (c.1588G> A (p.Gly530Ser) и c.2852A> G (p.Asn951Ser)). На основании проведенных дополнительных исследований авторами было установлено, что обе эти редкие мутации могут наследоваться лишь по линии отца, что объясняет отсутствие внечерепных проявлений у матери. В свою очередь сочетание мутаций в гене коллагена COL5A1 с мутацией ANO5 может объяснять более тяжелые проявления патологии у обоих сибсов, а также вариабельность клинических проявлений, не полностью укладывающихся в классическую картину НО, GC и GDD [3].
Полученные данные позволили исключить данную семью из расчетов отягощенности НО. В результате общая распространенность составила 1:18424, отягощенность – 5,43.
Заключение
В результате проведенного генетико-эпидемиологического исследования и приведенного клинического примера показано изменение показателей отягощенности и распространенности (с 1:16582 и 6,03 до 1:18424 и 5,43) после адекватной дифференциальной диагностики всех случаев заболевания, а также необходимость по возможности полного обследования всех доступных для осмотра членов семьи пробанда с использованием современных методов исследования, в том числе молекулярно-генетических исследований (включая полноэкзомные исследования), для точной постановки диагноза, определения прогноза и тактики возможного лечения пробанда, корректного медико-генетического консультирования.
Исследование выполнено при частичной финансовой поддержке РФФИ в рамках научных проектов №17-04-00288, РНФ 17-15-01051.