



Болезнь Альцгеймера (БА) признана Всемирной организацией здравоохранения глобальной приоритетной общественной проблемой. Несмотря на большие успехи в вопросе понимания патогенеза БА и его концептуализации, с того времени как Алоис Альцгеймер [Alzheimer, Alois] в 1917 г. впервые зафиксировал первый случай заболевания [1], до сих пор адекватной терапии данной формы патологии найти не удалось.
Эпидемиология
Деменция – приобретенное прогрессирующее когнитивное нарушение, которое значительно ухудшает качество жизни, является одной из основных причин инвалидности и приближает летальный исход. По текущим оценкам, 44 млнчеловек в настоящее время страдают деменцией. Прогнозируется, что их число возрастет более чем втрое к 2050 г., тогда как ежегодная стоимость лечения деменции только в США превышает 600 млрд долларов [2]. В Англии и в Уэльсе деменция является главной причиной смертности в целом, ее доля составляет 11,6% от общей смертности, зарегистрированной за 2015 г. Недавние исследования показывают, что в западных странах заболеваемость деменцией, особенно среди мужчин, имеет тенденцию к снижению. И хотя неясно, какие причины это вызывают, предполагается, что это может быть связано с регуляцией сосудистого риска. В ближайшие годы увеличение распространенности деменции ожидается в странах с низким и средним уровнем дохода, где отмечензакономерный рост сердечно-сосудистых заболеваний, артериальной гипертензии и сахарного диабета. БА является основной причиной слабоумия, на ее долю приходится 50–75% всех случаев его развития, и в первую очередь это – лица пожилого возраста, количество которых удваивается каждые 5 лет после 65 [2].
Этиология
Несмотря на то, что подавляющее большинство форм заболевания являются спорадическими, мутация в трех генах – белке-предшественнике амилоида (APP), пресенилине 1 (PSEN1) и пресенилине 2 (PSEN2) – вызывает редкую (<0,5%) семейную форму БА (fAD). Ее симптомы развиваются раньше, чем при спорадической БА, обычно в возрасте между 30 и 50 годами [3].
«Типичное» позднее начало БА, вероятно, обусловлено комплексным взаимодействием генетических факторов и факторов окружающей среды. В настоящее время считается, что приблизительно 70% риска развития БА связано с наследственностью. Ген аполипопротеина E (APOE), который имеет три варианта: e2, e3 и e4, обусловливает высокий риск развития спорадической БА. По сравнению с носителями non-e4 гетерозиготыe 4 имеют отношение шансов развития БА, равное 3, увеличиваясь до 12 в гомозиготах. Общегеномная ассоциация провела исследования с использованием нескольких тысяч образцов, выявив более 20 генетических факторов риска, связанных с воспалительными процессами, метаболизмом холестерина и рециркуляцией эндосомальных путей [4]. В частности, в настоящее время считается, что активация микроглии в ответ на отложение амилоида играет ключевую роль в патогенезе БА. Каждый из этих относительно распространенных вышеперечисленных генов несет очень небольшую вероятность развития патологии, но их объединение при полигенном типе наследования может увеличить риск возникновения заболевания [5]. Исследования с использованием генетического секвенирования позволили выявить ряд других редко встречающихся генов, обусловливающих относительно высокий риск развития БА, которые в свою очередь также дают представление о происхождении нейродегенеративного заболевания.
Эпидемиологические данные свидетельствуют о том, что наличие высокого уровня образования и занятия спортом могут снизить риск развития БА, в то время как сахарный диабет и гипертония в среднем возрасте повышают этот риск. Ожирение долгое время считалось фактором риска развития деменции и БА, но недавно этот факт был поставлен под сомнение [6]. Механизмы, посредством которых сосудистые факторы риска могут повлиять на развитие БА, остаются неясными, не в последнюю очередь потому, что лишь немногие эпидемиологические исследования имеют патоморфологическое подтверждение диагноза. Сосудистые факторы риска могут увеличить вероятность возникновения клиники БА посредством «двойного удара» – за счет цереброваскулярного повреждения; в иных случаях сосудистое повреждение может напрямую влиять на развитие БА.
Патоморфология
Кардинальным патоморфологическим признаком БА является наличие амилоидных бляшек и нейрофибриллярных клубков(NFTs). Кроме того, наблюдаются нейропильные нити, дистрофические нейриты, ассоциированный астроглиоз и активация микроглии, а также церебральная амилоидная ангиопатия. Последствиями этих патологических процессов в дальнейшем будет являться нейродегенерация с потерей синапсов и нейронов, что приведет к макроскопической атрофии. Смешанная патология особенно часто встречается у пожилых людей и включает сосудистые заболевания и наличие телец Леви. В самом деле, даже при семейной форме БА (fAD) часто встречаются тельца Леви, механизм формирования которыхостается неясным [7].
Амилоидные бляшки представляют собой внеклеточные скопленияамилоида, в основном состоящие из аномально свернутых Aβ белков с 40 или 42 аминокислотами (Aβ40 и Aβ42) – двумя побочными продуктами метаболизма APP. Белок Aβ42 в бляшках является более распространенным, чем Aβ40, из-за более высокой скорости фибриллирования и нерастворимости. Отложение амилоида не всегда сопряжено с прогрессированием заболевания, хотя широко распространено в неокортексе. Недавно стало известно, что амилоид оказывает влияние и на подкорковые структуры. В отличие от нейрофибриллярных клубков, амилоидные бляшки в меньшей степени вовлекают энторинальную кору и гиппокамповую формацию [8]. Различные системы стадирования Aβ включают систему Браака (Braak), критерий Тала (Thal) и Консоциума по созданию реестра для болезни Альцгеймера (the Consortium to Establish a Registry for Alzheimer Disease, CERAD) [8].
Нейрофибриллярные клубки в основном состоят из парных спиральных нитей, образованных гиперфосфорилированным тау-белком. Патология тау-белка обычно начинается в аллокортексе медиальной височной доли (энторинальная кора и гиппокамп), затем распространяется на ассоциативный изокортекс. Первичные сенсорные, моторные и зрительные области в основном не страдают. Нейрональные и синаптические потери обычно параллельны образованию нейрофибрилл. Таким образом, клинические признаки и тяжесть БА лучше коррелируют с патологией NFT, в то время как β-амилоидная патология служит индикатором патофизиологического процесса еще до появления клинических симптомов и на ранних стадиях заболевания [9].
Было предложено несколько патологических критериев для диагностики БА. Ранние попытки использования либо амилоидных бляшек, либо NFTв качестве диагностических критериев были ограничены их низкой специфичностью и чувствительностью [10]. Предыдущие патологические критерии БА Национального института по проблемам старения (National Institute on Aging, NIA) и Института Рейгана объединяли оценку невритных бляшек Консоциума по созданию реестра для болезни Альцгеймера (CERAD) с системой Braak и стадией NFTBraak, получив три диагностических категории вероятности возникновения заболевания: высокую, среднюю и низкую. Соответственно, диагноз БА может быть поставлен, только если критерии высокой или средней вероятности БА сочетаются с клиникой слабоумия (деменции). Использование клинико-патоморфологического подхода приводило к тому, что установление достоверного диагноза было возможно только посмертно или после проведения биопсии головного мозга, использование которой крайне ограничено. В обновленных рекомендациях Национального института по проблемам старения и Альцгеймеровской ассоциации (NIA-AA) предпринята попытка решить эту проблему, признавая вероятность разрыва между клиническими проявлениями и патоморфологическими процессами, лежащими в их основе[11]. Было постулировано, что если прижизненно обнаруживаются типичные патофизиологические признаки, отражающие патоморфологические изменения альцгеймеровского типа, то можно диагностировать заболевание на ранних стадиях.
Патогенез
Амилоидная гипотеза – распространенная теория патогенеза БА – предполагает, что накопление патологических форм Aβ, продуцируемых за счет последовательного расщепленияAPP ферментами β- и γ-секретазой в головном мозге, является первичным патологическим процессом, вызванным дисбалансом между продукцией и клиренсом Aβ. Считается, что формирование NFT и последующие нейронная дисфункция и нейродегенерация, которая в свою очередь, возможно, опосредована воспалением, являются взаимосвязанными процессами [12] (рисунок). Влияние Aβ обусловлено функционированием генетического нейронального аппарата: все мутации, связанные сfAD, участвуют либо в генерации, либо в процессинге Aβ и приводят к относительному перепроизводству токсичных форм β-амилоида. И наоборот, миссенс-мутация (точечная) APP (A673T) в течение всей жизни приводит к снижению расщепления APPβ-секретазой, что снижает клинический риск развития БА [13]. При спорадической болезни аполипротеин E (ApoE) участвует в клиренсе амилоида, как и многие другие гены риска.
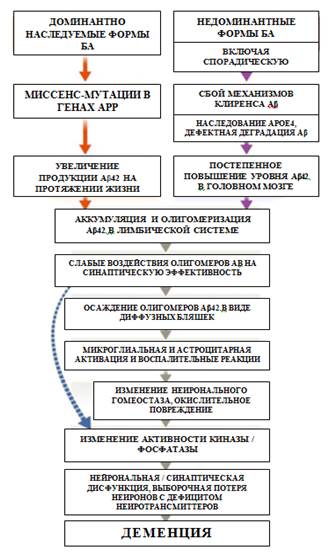
Основные патогенетические процессы, приводящие к БА. Изогнутая синяя стрелка указывает, что Аβ олигомер может приводить к синаптическим и невритическим повреждениям и вызывать тау-гиперфосфорилирование как дополнение к активации разрушительного воспалительного каскада [14]
Хотя первоначально считалось, что фибриллярный амилоид в бляшках с плотным ядром является критическим для развития БА, сейчас полагают, что растворимые олигомеры Aβ могут быть наиболее патологическими формами: олигомеры, применяемые к нейронам invitro, вызывают синаптическую дисфункцию, повреждают дендритные шипы и приводят к гибели нейронов. Олигомеры человека индуцируют гиперфосфорилирование тау-белка в эпитопах, связанных с БА, и вызывают нейритную дистрофию в культивируемых нейронах. Таким образом, бляшки могут действовать как «резервуар» при диффундировании олигомеров или способны выступать в качестве защитного механизма, связывающего токсичныйАβ, пока не будет достигнута точка физиологического насыщения [15].
Хотя наличие скопления Aβ необходимо для верификации диагноза БА, тот факт, что значительная часть пожилых людей умирает с доказательно присутствующим отложением Aβ без нарушений когнитивных функций, показывает, что этого недостаточно для развития деменции при БА.
Установлено, что соотношение Aβ-растворимый олигомер /бляшка ниже у пациентов с бессимптомным амилоидозом, чем у пациентов с БА. Данный факт согласуется с концепцией, полагающей, что бляшки могут выступать в качестве защитного резервуара. Образование тау-белка является неотъемлемой частью патогенеза БА, о чем говорит требование наличия не только Aβ, но и тау-белка для диагностики БА. Кроме того, прослеживается тесная связь между степенью нейродегенерации и количеством тау-белка в нейрональной ткани. Тем не менее, хотя мутации в гене тау-белка приводят к накоплению тау-белка и различным нейродегенеративным деменциям фронтотемпорального спектра, в отличие от мутации в β-амилоидных генах, одиночные мутации, кодирующие образование тау-белков, не вызывают БА [16].
Обнаружение при БА биомаркеров Aβ в цереброспинальной жидкости (ЦСЖ), изменений при позитронно-эмиссионной томографии (ПЭТ) и накопления патологического тау-белка привело к многочисленным исследованиям, изучающим прогресс и взаимодействие между этими процессами внутри организма (in vivo). Исследования на здоровых пожилых людях и пациентах со спорадической БА и fБА предоставляют дополнительные доказательства того, что амилоидная патология начинает развиваться за много лет до появления клинических симптомов. Она предшествует изменениям в ЦСЖ, на ПЭТ и, наконец, опережает развитие клинических симптомов. В то же время имеются данные, позволяющие предположить, что накопление β-амилоидаявляется более значимым, чем синтез тау-белка при БА. У некоторых здоровых пожилых людей обнаруживается патологический тау-белок без β-амилоида, что может быть частью закономерного процесса старения, а значит, не отражает нейродегенерацию, развивающуюся при БА [17].
Существует большой интерес к механизмам, с помощью которых белки БА синтезируются в определенных областях мозга и распространяются в другие его отделы. Аномальная конформация Aβ и тау-белка способна вызвать конформационные изменения в структурно нормальном пептиде так же, как это происходит при прионной болезни, при которой может быть цепная реакция трансформации пространственной структуры белков, затрагивающая не один нейрон. Очаг изначального патологического процесса можно будет определить по тому, какие корковые сети подвержены воздействию, приводящему к их распаду, что и объясняет фенотипическое разнообразие, наблюдаемое при БА. Известно, что амилоидная и тау-патологии имеют решающее значение в патогенезе БА, но как они связаны, до сих пор неясно. Можно привести ряд доказательств, говорящих, что врожденная иммунная система играет не последнюю роль в патогенезе БА. Результаты посмертной биопсии демонстрируют соседство активной микроглии с амилоидными бляшками. Ряд генов, кодирующих высокий риск возникновения БА, в том числе CR1, CD33 и TREM2, участвуют в иммунных процессах [18]. Клинические исследования с использованием ПЭТ лигандов, которые связываются с активированной микроглией, свидетельствуют о важной роли воспалительных изменений в нервной тканипри БА. Вопрос о том, является ли нейровоспаление защитным или повреждающим механизмом, на данный момент остается открытым [19].
Заключение
Таким образом, анализ научной литературы, посвященной БА, позволяет заключить, что до сих пор нет единой теории, объясняющей возникновение и прогрессирование данного заболевания. Дальнейшие исследования в данной области, возможно, приведут к формированию новой концепции патогенеза БА.